Increased detection sensitivity of MS machinery coupled with high-powered peak recognition software has improved the ability to isolate, monitor, and analyze substance compositions of complex samples in very minute detail. Using these technological advancements, recent studies on peptidoglycan composition have begun to use automated LC-MS feature extraction techniques12,13,14,24 over older HPLC-based methodology11,25,26,27,28,29,30,31. Although there are numerous generic feature extraction software packages available, commercial software using recursive feature extraction is rapid and highly robust by automatically identifying and combining all the charges, isotopes, and adduct versions of each muropeptide found within the LC-MS dataset (Figure 3). In addition, initial retention times, m/z and isotopic patterns of extracted features are used to reassess (recursive) the dataset to ensure accurate identification of each feature in all data files. Therefore, the recursive algorithm aids in validating and increasing confidence in peak identification. Most generic feature extraction programs do not group charges/isotopes, etc. and will require this as an additional manual step. In addition, generic programs will be less robust as features are extracted separately within each data file and not as an entire dataset, which is part of the recursive algorithm.
The peptidoglycomic protocol presented here was recently used to examine the compositional changes of PG between two physiological growth conditions, namely, free-swimming planktonic and stationary communal biofilm12. Using a highly sensitive QTOF MS coupled with the recursive feature extraction, 160 distinct muropeptides were recognized and tracked. This represented eight times the number of muropeptides identified in this organism previously29,32, and greater than double the muropeptides identified using other methodologies in other organisms10,14,24.
Associating each m/z peak extracted from the MS data with a particular muropeptide is facilitated by cross-referencing with a database of known and predicted muropeptide structures. The fragmentation MS/MS chromatogram (Figure 4) for each extracted feature is compared to the fragmentation profile (Figure 4, gray inset) of the muropeptide proposed using the database.
Peptidoglycomic data can be viewed in a number of different ways depending on the experimental setup and the questions being asked. Such graphical analysis can include principal components analysis (PCA), scatterplots, volcano plots, heat maps, and hierarchical clustering analysis. For example, volcano plots highlight muropeptides that demonstrate a statistically significant high magnitude of abundance change between the tested conditions (Figure 5A). These selected muropeptides which represent significant abundance changes between the tested conditions can be further examined for muropeptide modifications. These modifications can include the presence of amino acid substitutions, acetylation changes, or the presence of amidase activity. When examined together, multiple muropeptides possessing the same modification can be examined for a trend toward one experimental condition (Figure 5A—highlighted points green) and the entire group assessed for significance (Figure 5B). Tracking a muropeptide modification in this way, can indicate a particular enzymatic activity that is affected by the experimental parameter. In addition, outliers from this trend may indicate enzymatic activity with a particular specificity or biological function (Figure 5A—highlighted points orange).
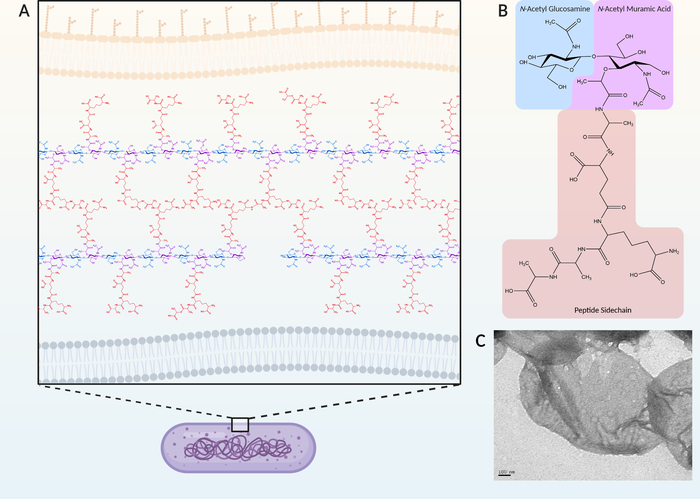
Figure 1: Example of a typical Gram-negative peptidoglycan structure. (A) In Gram-negative bacteria, peptidoglycan is located in the periplasm between the inner and outer membranes. (B) A single muropeptide consists of a β-1,4-linked N-acetyl glucosamine (GlcNAc) (blue) and a N-acetyl muramic acid (MurNAc) (purple) with an appended peptide sidechain (orange). The peptide sidechain can be crosslinked to the sidechain of adjacent muropeptide producing the mature mesh-like peptidoglycan (A). Purification involves the isolation of the peptidoglycan from the entire cell as a sacculus where all other cellular material has been stripped away. (C) Transmission electron micrograph of a peptidoglycan sacculi. In comparison, Gram-positive PG can consist of a greater array of variations in structure and is part of Gram-positive taxonomic classification33. Please click here to view a larger version of this figure.
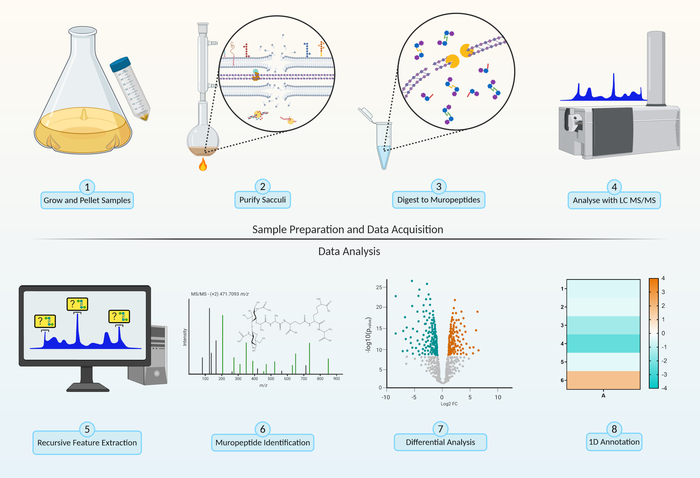
Figure 2: Peptidoglycomics workflow. Sample Preparation. Step 1, grow and pellet bacterial cells (section 1.1). Step 2, purify peptidoglycan sacculi by 4% SDS boil (section 1.2). Data Acquisition. Step 3, enzymatic digestion of sacculi to produce muropeptides by breakage of the β-1,4-linkage between the N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) of the peptidoglycan backbone (section 2.1). Step 4, analysis of muropeptide intensity through LC-MS/MS (section 2.2). Data Analysis. Step 5, recursive feature extraction identifies and collect all charges, adducts and isotopes associated with a single muropeptide (section 3.1). Step 6, identification of muropeptides by comparing predicted fragmentation with MS/MS chromatograms (section 3.3). Step 7, bioinformatic differential analysis (section 3.2) comparing peptidoglycan compositional changes between different experimental parameters. Step 8, examine the global change in muropeptide modifications within the different experimental parameters using 1D annotation (section 3.4). Please click here to view a larger version of this figure.
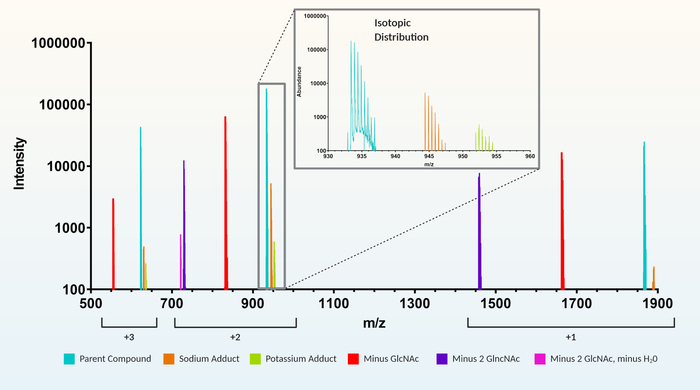
Figure 3: Example of a recursive feature extraction. For a muropeptide representing a peptide sidechain of alanine (A), iso-ᴅ-glutamate (E), meso-diaminopimelic acid (m), alanine (A) crosslinked to the AEmA of the adjacent muropeptide sidechain (1864.8 m/z). Included in the extracted feature for 1864.8 m/z are charges (+1, +2, and +3), adducts (e.g., sodium and potassium), loss of GlcNAc (1 or 2 GlcNAc), and multiple isotopic peaks for each variation (e.g., zoomed inset). Please click here to view a larger version of this figure.
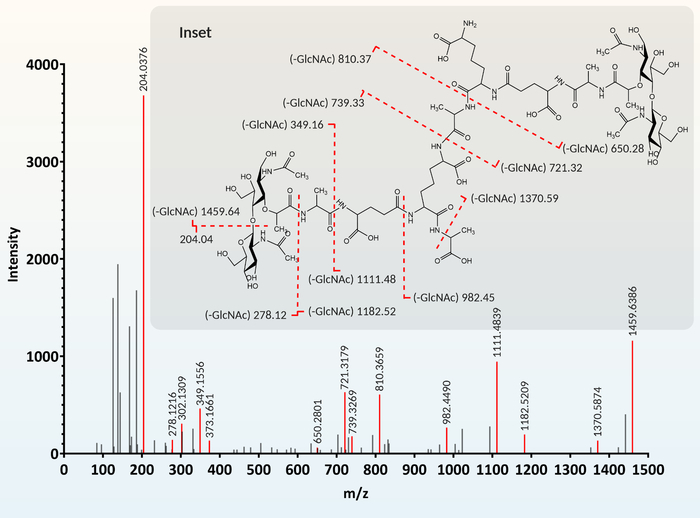
Figure 4: Muropeptide fragmentation and identification. For annotation, each m/z peak (feature) extracted from the MS chromatogram is given a proposed muropeptide structure based on similarity to a muropeptide library. To confirm this proposed structure, predicted MS/MS fragments are generated using a chemical drawing program (gray inset). This predicted fragmentation is compared to the MS/MS chromatogram. When predicted fragments (gray inset) match the MS/MS chromatogram, the proposed muropeptide structure is confirmed. The figure was modified from Reference12. Please click here to view a larger version of this figure.
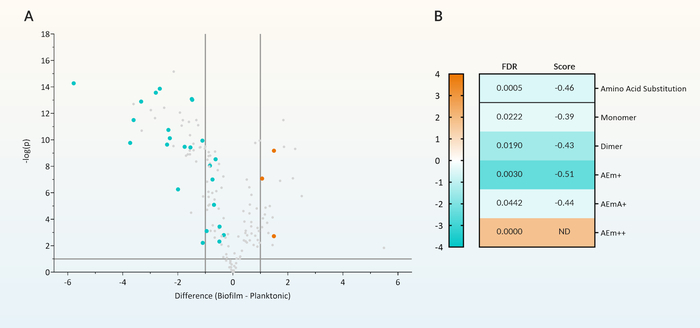
Figure 5: Differential analysis of peptidoglycan composition. (A) Volcano plot of the fold change and statistical significance of changes in muropeptide intensity between peptidoglycan purified from P. aeruginosa grown as either free-swimming planktonic or stationary biofilm culture. All muropeptides that have a modification that represented a change in the typical amino acid arrangement within the peptide sidechain are highlighted. Amino acid substituted muropeptides that showed a trend towards decreased abundance in biofilm-derived peptidoglycan are highlighted in green. Amino acid substituted muropeptides that were outliers to this trend and showed increased abundance in biofilm-derived peptidoglycan are highlighted in orange. (B) Heat map of the global fold change in abundance of all the amino acid substituted muropeptides with increased abundance (orange) and decreased abundance (green) in biofilms. These muropeptides were regrouped and assessed for whether amino acid substitution occurred on monomers, crosslinked dimers, or whether the fourth (AEm+), fifth (AEmA+) or both amino acids (AEm++) were substituted. The significance of each group of muropeptides were assessed by 1D annotation with FDR < 0.05 for significance and the associated 1D score is displayed. 1D annotation can only be performed on more than 2 muropeptides (e.g., AEm++ substitution was only found on two muropeptides). Therefore, in this case, significance must be examined for the individual muropeptides and not on the group. The figure was modified from Reference12. Please click here to view a larger version of this figure.
