All animal work was approved by the Ethical Committee N 59 and the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche under the file number APAFIS#15859-2018051710341011v3. Some of the steps described below are specific to our equipment and software but could be easily adapted to different equipment.
1. Injection preparation
- Prepare 75 mL of 1% agarose solution in Embryo Medium (EM).
- Place the injecting mold in a 90 mm Petri dish and pour approximately 50 mL of agarose, enough for the mold to float. Let the agarose solidify and remove the injecting mold.
- Prepare an agarose-coated dish by pouring 1 mL of agarose in a 30 mm Petri dish.
- Prepare 4 µL of 30 ng/µL Histone2B-mCherry mRNA solution by diluting the stock solution in RNase-free water and keep on ice.
NOTE: Take care to wear gloves while manipulating mRNA to avoid RNase-mediated degradation. - Pull an injection needle from a capillary using the micropipette puller.
2. Embryo preparation
- Once fishes have laid eggs, collect, rinse, and harvest in a 90 mm Petri dish in EM. Place the embryos in a 28.5 °C incubator.
- Wait 20 min for the first cell to become visible.
- Transfer 30 embryos to the injection plate filled with EM. Squeeze embryos in the grooves using slightly blunt forceps and orient them with the animal pole up.
- Using a microloader tip, fill an injection needle with 2 µL of mRNA solution. Insert the needle in the capillary holder placed in a micro-manipulator connected with polytetrafluoroethylene (PTFE) tubing to an air injector.
- Under the stereomicroscope, carefully break the tip of the needle.
- Inject the mRNA solution in the 1-cell stage embryos by inserting the needle in the cell.
NOTE: The volume injected is approximately one-third of the cell volume. - Place back injected embryos in the 28.5 °C incubator.
3. Preparation of the two-photon microscope
NOTE: Two lasers are used in this protocol. One is used to image GFP (at 920 nm) and perform ablations (at 820 nm). It will be referred to as the green/ablation laser. The other is used at 1160 nm to image mCherry. It will be referred to as the red laser.
- Set the green/ablation laser to 820 nm (ablation wavelength) and the red laser to 1160 nm (mCherry excitation).
- Using movable mirrors on the optical path, align green/ablation and red laser beams both at the entry and exit of the scan head.
NOTE: This increases the laser beam focus and minimizes focal volume for excitation and ablation. - Measure the maximum power of the green/ablation laser at 820 nm under the objective. To do so, place the power meter under the objective, close the black chamber, set green/ablation laser power to 100%, and open the shutters. Compute the percentage of laser power needed to reach 300 mW.
- Set back the green/ablation laser to 920 nm (GFP excitation) and set the laser power to 7%. Set the red laser power to 15%.
- Activate epi-PhotoMultiplier Tubes (PMT) detectors for green and red lines; set green and red line PMT sensitivity to 65.
- Set the field of view to 400 x 400 µm, image resolution to 512 x 512 pixels, and scanning frequency to 800 Hz.
- Select 3D Timelapse Imaging mode. Then, create a folder and activate Autosave for data after each acquisition.
- Assemble the heating chamber and set it to 28 °C. Wait at least 10 min for the chamber and the objective to warm.
4. Mounting the embryo
- Under a fluorescence stereomicroscope, identify embryos at 70% epiboly that express GFP.
NOTE: Select embryos with a bright signal in the axial mesoderm and no background fluorescence for better imaging quality. - Transfer three to four selected embryos in the agarose coated dish (step 1.3) using a plastic Pasteur pipette and carefully dechorionate them using fine forceps.
NOTE: Dechorionated embryos are very delicate and will burst upon contact with air or plastic. - Pour 1 mL of 0.2% agarose in 1x penicillin-streptomycin EM in a small glass vial. Place the vial in a preheated 42 °C dry block heater.
NOTE: The following steps must be performed quickly to allow embryo orientation before agarose sets. - Transfer a dechorionated embryo in the 0.2% agarose glass vial using a fire-polished glass pipette. Take care not to add too much EM in the agarose to avoid diluting it. Discard the remaining EM from the pipette and aspirate the embryo back along with enough agarose to cover the slide of the glass bottom dish before the embryo falls out of the pipette.
- Blow the agarose and the embryo on the glass slide of the dish. Take care not to let the embryo touch the air or the plastic side of the dish. Next, fill the chamber around the glass slide with agarose.
- Use an eyelash to orient the embryo so that the targeted region is at the top (Figure 1B).
NOTE: When orienting embryos, take care to only touch the blastoderm, not the very fragile yolk. Agarose will set in around 1 min, depending on room temperature. - Wait ~5 min for the agarose to set completely, and then add a few drops of penicillin-streptomycin EM.
5. Locating the embryo and pre-ablation imaging
- Place the glass bottom dish under the objective in the heated chamber. Immerse the objective in penicillin-streptomycin EM and close the heated chamber.
- Move the slider to set the light path to oculars. Then, using oculars, fluorescent lamps, and stage control, find an embryo and set the focus to the surface of the embryo.
- Turn the fluorescence lamp off, set the light path to PMTs, and close the black chamber.
NOTE: Be careful to turn off all light sources in the black chamber as it might damage the PMTs. - Start live imaging and locate axial mesoderm. Adjust the green/ablation and red laser powers to have a good signal (i.e., between 1,000 and 20,000 photons per pixel for GFP expressing areas). Use the red channel to move the stage to the very top of the embryo and set this position as Z = 0.
- Choose a time-step of 1 min and a Z-step of 2 µm. A Z-course of 110 µm is sufficient to encompass the whole polster and is acquired in less than 1 min with these settings. Set the first slice 15 µm above the axial mesoderm (in the more superficial ectoderm).
NOTE: The polster moves along a curved line so that the bottom slice of the Z-stack should be set 30 µm deeper than the polster deepest position to accommodate its movement during the time-lapse imaging (Figure 1E). - Record 10-15 min of pre-ablation movie.
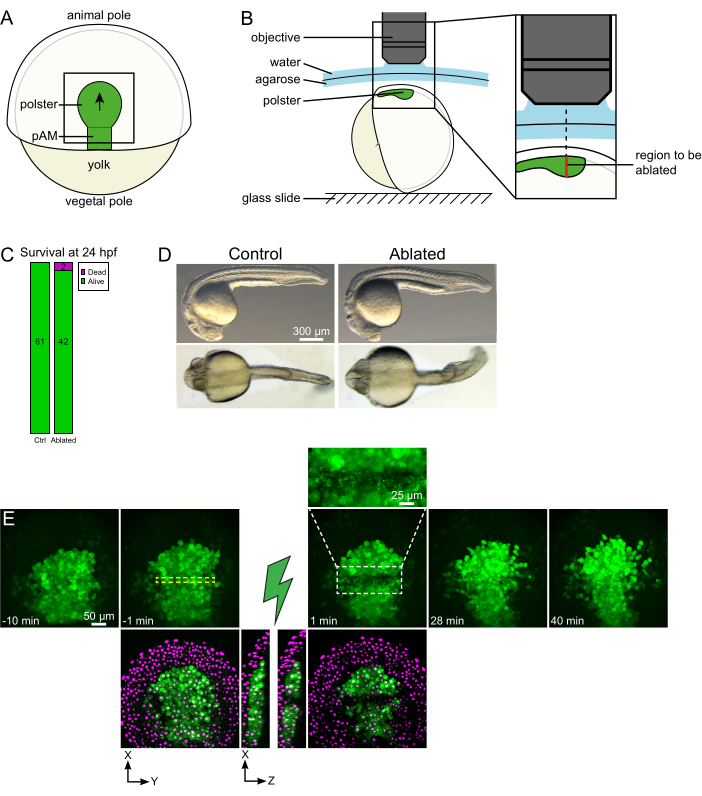
Figure 1: Successful outcome of laser ablations. (A) Scheme of a gastrulating embryo at 70% epiboly in dorsal view; pAM: posterior axial mesoderm; black arrow marks the direction of polster migration; black square indicates a typical field of view for ablations in the polster. (B) Scheme of embryo mounting for polster severing. Lateral view. The embryo is mounted such that the plane of the polster is perpendicular to the optical axis. (C) survival and (D) morphology of control and ablated embryos at 24 h post-fertilization. Scale bar is 300 µm. (E) Time sequence from laser ablation in the polster of a Tg(gsc:GFP) embryo expressing Histone2B-mCherry. Views with the green channel only are maximum projections. The close-up displays the ablated area containing cell debris. Views with green and red (displayed as magenta) channels are XY and XZ slices before and after ablation (the green lightning bolt represents ablation). XZ slices show that the overlying tissues (magenta nuclei without GFP expression) have not been affected by the ablation of underlying structures. The yellow dashed box corresponds to the ROI selected for laser ablation treatment. The scale bar is 50 µm in large views and 25 µm in the close-up. Please click here to view a larger version of this figure.
6. Target location and laser ablation
- Locate the polster contour on live imaging and, using the Electro-Optic Modulator Region of Interest (EOM ROI) tool, draw a 20 pixel (15 µm) large rectangle that spans the width of the polster. Place this rectangle in the middle of the polster (Figure 1E).
- Note the axial position of the highest and lowest planes containing polster cells. Ablations will be performed every 10 µm in between these two planes. Take care that the ROI does not overlap the yolk cell on any of these planes.
- Place the stage at the lowest Z position of the interval. Ablations must be performed bottom-up as debris absorb light.
- Set the green/ablation laser wavelength to 820 nm and set the Power Percentage to obtain an exit power of 300 mW (step 3.3).
- Set the Imaging Frequency to 200 Hz.
- Set green/ablation laser imaging EOM to 0 and select ROI-Treat mode.
- Turn on the EOM and set the treatment to start immediately (after 0 frame).
- Set the Imaging Mode to Timelapse and de-activate Autosave.
- Set the Time Step to Fast mode.
- Set the Number of Treatment Frames and Number of Frames to the value corresponding to the targeted depth (Table 1).
| Depth (µm) | Treatment frames |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
Table 1: Suggested number of laser treatment frames as a function of targeted cell depth in the embryo (0 being the embryo's surface).
- Start imaging. The acquisition is black as the shutter to PMT closes during EOM treatment.
- Move up the stage to the next Z position of the list (step 6.2).
- Repeat steps 6.10 to 6.12 until the top of the polster is reached.
7. Post-ablation verification and imaging
- Set the green/ablation laser to 920 nm and 5% power. Set the green/ablation laser imaging EOM to 100 and select the Fullfield mode.
- Set the Imaging Frequency to 800 Hz. Turn EOM off.
- Go through the whole stack in live mode to check whether every plane has been ablated. If this is not the case, go back to step 6.2.
NOTE: Ablation sometimes induces a vertical shift of neighboring tissues so that the Z-stack might have to be redefined. - Set the Imaging Mode to 3D Timelapse and re-activate Autosave. Record 40-60 min of post-ablation movie.
- Check, in the post-ablation movie, whether the targeted cells were effectively ablated. Fluorescence recovery, or targeted cells occupying space and preventing follower cells from moving through, indicate that targeted cells were only photobleached and not ablated (Figure 1E and Figure 2A).
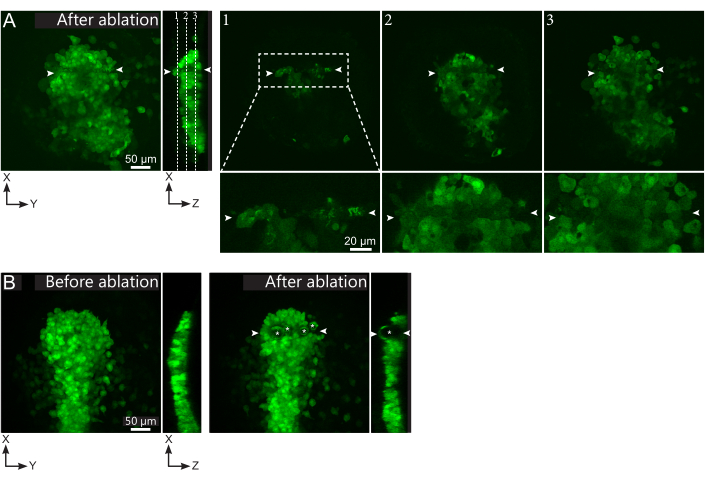
Figure 2: Negative results of laser ablations. (A) Typical examples of potential failures in laser ablation. Large XY views are maximum projections, XZ view is a reconstructed section. Laser treated area is located between the two white arrowheads. Three focal planes are highlighted in the reconstructed section and displayed on the right. They correspond to three different kinds of failures. Plane 1 shows that cells above the polster have been ablated. This can be identified by the presence of autofluorescent debris on this focal plane (see close-up) above the polster (see position of plane 1 on the reconstructed section). This likely results from an incorrect definition of the region to be ablated. Plane 2 shows cells that have been bleached but not ablated. They can be identified as the low fluorescence signal still reveals intact cell contours (see close-up). Plane 3 displays intact cells, which have hardly been bleached by laser treatment. This could result from an incorrect definition of the region to be ablated or from poor treatment. In the situations depicted in planes 2 and 3, it is possible to re-apply the ablation treatment to the non-ablated targeted cells. The scale bar is 50 µm in large views and 20 µm in close-ups. (B) A typical example of bubbles (marked by white asterisks) formed by cavitation because of a too intense laser treatment. Such bubbles are not limited to a Z-plane, sometimes even spanning the full height of the polster, deforming neighboring tissues. The scale bar is 50 µm. Please click here to view a larger version of this figure.
8. Data analysis
- Open time-lapse series with the image analysis software and set correct pixel size.
- In the Spot function, set the Object Size to 10 µm, as this is the average nucleus size during gastrulation. Then, run the Spot function to detect and track the nuclei.
NOTE: Detection may be slightly improved by considering the lower axial resolution, fitting a 12 µm long ellipsoidal shape along the Z-axis. - Use filters to remove false positives. In the Tg(Gsc:GFP) line, cells from the embryonic axis and some endodermal cells are labeled in green. Hence, filtering on green intensity allows a quick selection of these cells (Figure 3A).
- Set the maximal distance between consecutive points to a value compatible with the speed of the cells.
NOTE: Be careful to consider the time interval between two frames. Polster cells migrate at 2.8 ± 0.8 µm/min. Hence, allowing 4 µm of maximum displacement for a time step of 1 min removes most artefactual tracks. - Allowing gaps over one or two time points provides longer continuous tracks but may introduce tracking errors. If a nucleus is not detected correctly at a one-time point, consider re-running spot detection with different parameters/filters.
- Visually check tracks and, if necessary, correct them.
- Export the results as a .xlsx file. Process the file using published spreadsheet routines24 (Figure 3B) and custom routines on data analysis software (available on request).
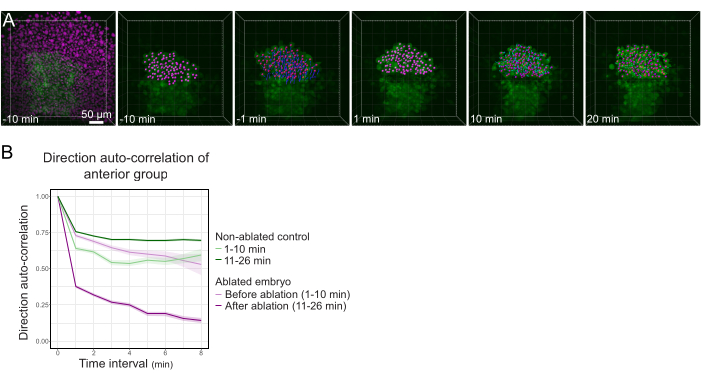
Figure 3: Isolation of the anterior half of the polster affects cell directionality. (A) 3D reconstructions a Tg(gsc:GFP) embryo expressing Histone2B-mCherry (displayed in magenta), before and after a laser ablation severing the polster in its middle. Nuclei belonging to the anterior half of the polster are marked with a magenta dot and tracked over time before and after ablation (see Movie S1). The scale bar is 50 µm. (B) As a measure of migration persistence, direction auto-correlation of cells belonging to the anterior part of the polster before and after ablation. Cells display a continuous motion before ablation, which drastically decreases after ablation, indicating loss of collective-oriented migration. Direction auto-correlation was also measured on cells forming the anterior half of the polster of a non-ablated embryo, as a control. The graph envelopes indicate standard error. Please click here to view a larger version of this figure.
