Selective Ribosome Profiling (SeRP) is the only method, to date, that captures and characterizes co-translational interactions, in vivo, in a direct manner1,2,3,4,5,6. SeRP enables global profiling of interactions of any factor with translating ribosomes in near codon resolution2,7.
The method relies on flash freezing of growing cells and preserving active translation. Cell lysates are then treated with RNase I to digest all mRNA in the cell except ribosome-protected mRNA fragments termed "ribosome footprints". The sample is then split into two parts; one part is directly used for the isolation of all the cellular ribosomal footprints, representing all ongoing translation in the cell. The second part is used for the affinity-purification of the specific subset of ribosomes associated with a factor of interest, for example: modifying enzymes, translocation factors, folding chaperones, and complex-assembly interactions. The affinity-purified ribosomal footprints are collectively termed the interactome. Then, the ribosome-protected mRNAs are extracted and used for cDNA library generation, followed by deep sequencing.
Comparative analysis of the total translatome and interactome samples allows for the identification of all orfs which associate with the factor of interest, as well as characterization of each orf interaction profile. This profile reports the precise engagement onset and termination sequences from which one can infer the decoded codons and the respective residues of the emerging polypeptide chain, as well as on the ribosome speed variations during the interaction7,8. Figure 1 depicts the protocol as a schematic.
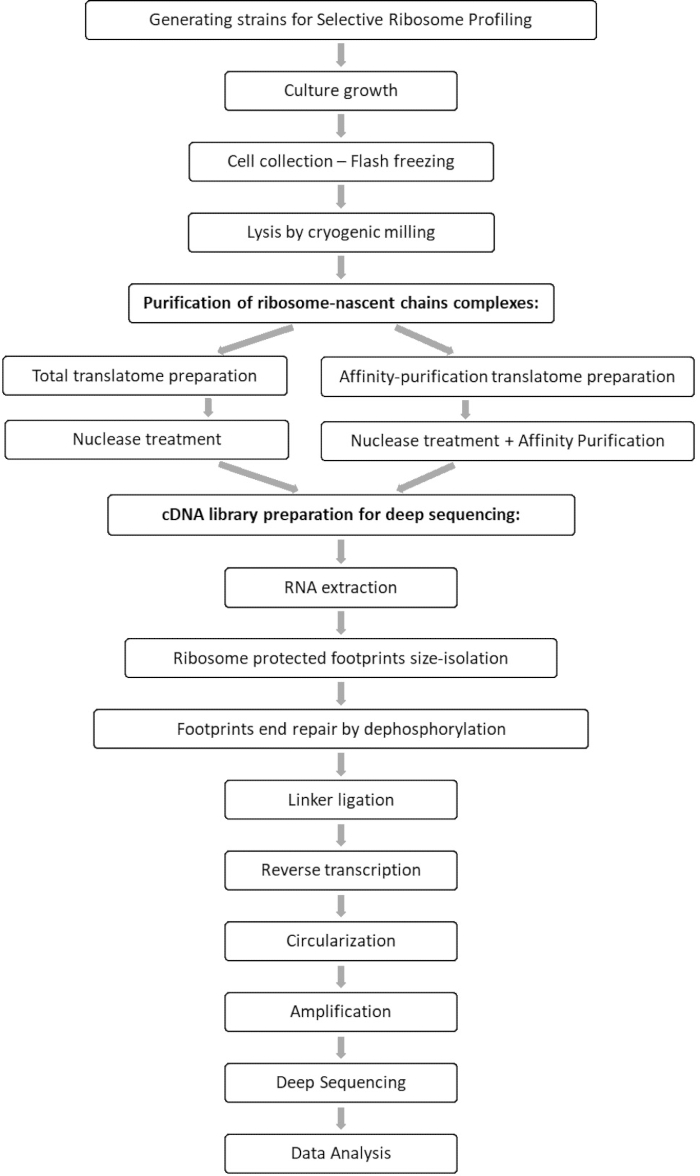
Figure 1: An overview of the SeRP protocol. This protocol can be performed in its entirety within 7-10 days. Please click here to view a larger version of this figure.
As illustrated in the flow chart of this protocol (Figure 1), cells were grown to log phase, and then collected swiftly by filtration and lysed by cryogenic grinding. The lysate was then divided into two: one for total ribosome-protected mRNA footprints and the other for selected ribosome-protected mRNA footprints, on which we performed affinity purification to pull-down the tagged protein-ribosome-nascent chains complexes. We ensured tagged protein expression and the success of the pull-down by western blot analysis, as can be seen in Figure 2. We validated the isolation of ribosome-protected footprints, which are typically 20-45 nt long by small RNA electrophoresis (2100 BioAnalyzer system), allowing for 5-10 nt shift in size detection, according to the system manual (Figure 3). Then, we generated a cDNA library for deep sequencing and big-data analysis. While generating the cDNA library, note that under-cycling can lead to low yield (as can be seen in lane 2 in Figure 3), but re-amplification is possible in order to recover the generated library. Over-cycling may occur when PCR primers are depleted but the reaction continues. When dNTPs are still present, the reaction proceeds, generating longer PCR artifacts with chimeric sequences due to PCR products priming themselves13 (as can be seen in lanes 3-4 in Figure 3, indicated by the visible smear). If the dNTPs' concentration also becomes limiting, products indicating the presence of heteroduplexes composed of only partially homologous library fragments can appear. Figure 4 acts as a reference, with lane 2 representing optimal amplification, and lane 3 an acceptable amplification. Samples from lanes 4 and 5 (cycles 10 and 11) should not be used due to the possibility of introducing PCR duplicates and artifacts. The generated library was further validated by high sensitivity DNA electrophoresis (the same BioAnalyzer system was used) for exact size distribution and quantification (Figure 5). After 3' end linker ligation, reverse transcription and PCR amplification, a cDNA length distribution as such is expected, with a sharp peak around 175 nt.
We trimmed and removed adapters and barcodes from the sequenced library, and only the reads between 20 and 45 nt were selected for further analysis. Figure 6 shows the resulting length distribution. The reads were divided into different groups of: coding sequences, introns, and intergenic sequences (Figure 7), and further classified as shown in Figure 8.
Final analysis for detection and characterization of co-translational interactions was performed based on the enrichment of ribosome-protected mRNA fragments, producing the graphs in Figure 9. We compared the normalized ribosome occupancy (at each nucleotide along each orf) of the total translatome to its corresponding selected translatome (nascent-interactome). Per nucleotide comparison eliminates translation rates artifacts. Reproducibility between biological replicates was evaluated by Pearson correlation (threshold > 0.6). We present Selective Ribosome Profiles, analyzing co-translational interactions of Vma2p with three proteins: the ribosome-associated chaperone Ssb1p, Pfk2p (Phosphofructokinase) and Fas1p (Fatty acid synthase), with each protein C` terminally tagged by GFP. We performed the protocol in biological replicates. Figure 9 A, D and G shows the experimental scheme of each affinity purification. We next show the ribosome occupancy of total translatomes compared to Ssb1 interactomes along the Vma2p orf, encoding for a subunit of the vacuolar H+-ATPase (Figure 9 B, E and H). Finally, we performed ratio-based ribosome-enrichment profiling (IP/Total) at each ribosome position in [codon/aa] along the orf (Figure 9 C, F and I). Comparing the co-translational interactions of these three proteins with Vma2p, which is being synthesized by the ribosome, revealed that Ssb1 chaperone engages the nascent Vma2p at four different regions along the orf, as we identified four significant enrichment peaks by SeRP. Differently, Pfk2p shows only one significant enrichment peak, as identified by SeRP, in a different position compared to the co-translational chaperone Ssb1. Analysis of Fas1 co-translational interactions with nascent Vma2p did not detect any significant enrichment. Thus, the comparison of these ratio-based enrichment ribosome profiles demonstrates this protocol`s power in detection and characterization of various co-translational interactions in near codon-resolution.
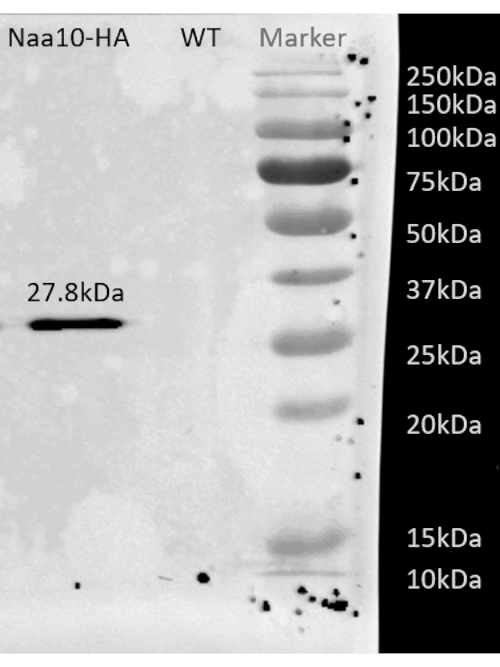
Figure 2: Representative western blot result after affinity purification of BY4741 strain with HA-tagged Naa10. Representative western blot result after affinity purification of BY4741 strain with HA-tagged Naa10 showing a band around 27.8 kDa, while the wild-type, as a negative control, shows no band. Please click here to view a larger version of this figure.
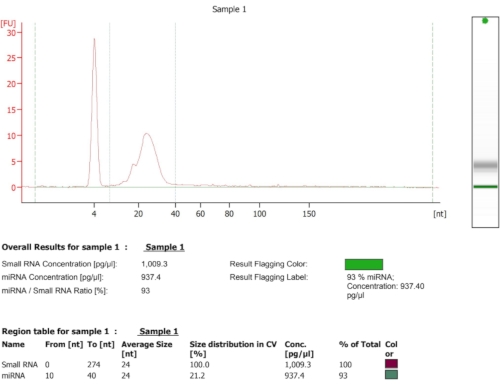
Figure 3: Representative BioAnalyzer result after footprint isolation and RNA extraction with acid-phenol:chloroform, and an average size of 25 nt. Please click here to view a larger version of this figure.
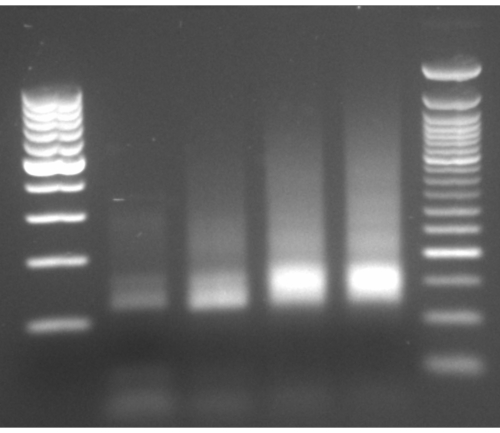
Figure 4: Representative gel-electrophoresis of PCR amplification. Representative gel-electrophoresis of PCR amplification with lanes 2-5 loaded with PCR products from cycles 8-11, and ladders on both sides. Please click here to view a larger version of this figure.
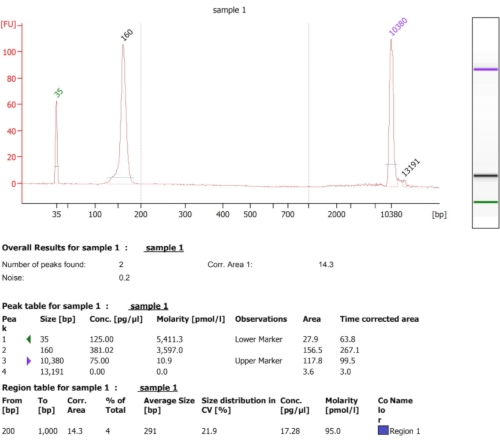
Figure 5: Representative BioAnalyzer result obtained following the creation of a cDNA library. Please click here to view a larger version of this figure.
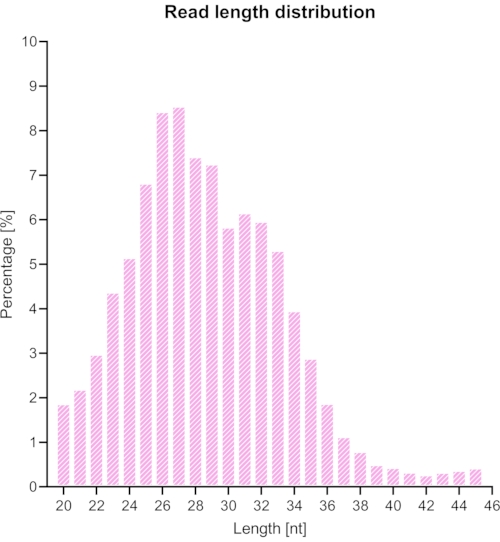
Figure 6: Expected length distribution of reads after the removal of the adaptors with Cutadapt (removing reads shorter than 20 or longer than 45). Please click here to view a larger version of this figure.
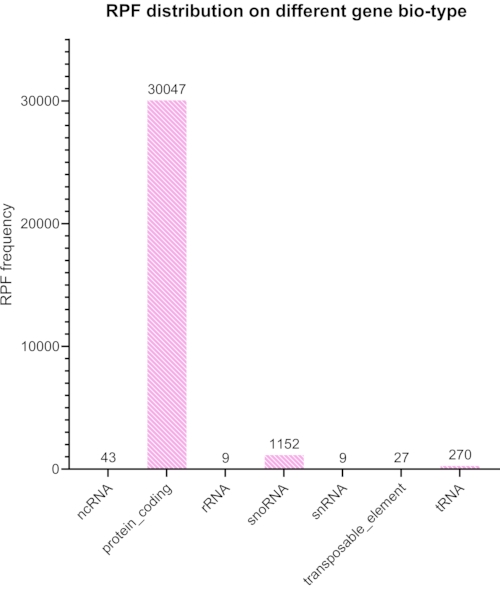
Figure 7: Expected alignment success percentage after removing non-coding RNA reads with Bowtie2 and using TopHat to align the remaining reads to different organisms. The sample was taken from S. cerevisiae (a mutated variant of BY4741). Please click here to view a larger version of this figure.
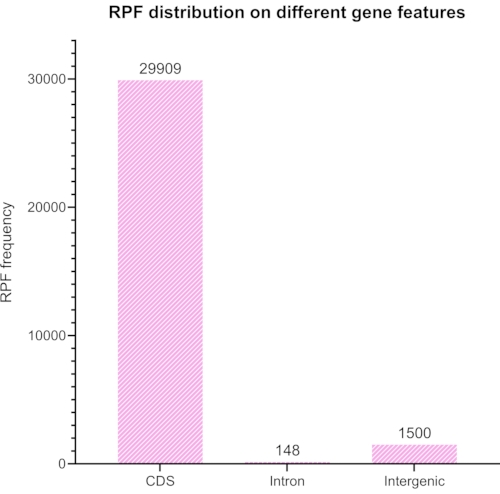
Figure 8: A graph generated with RiboToolkit representing expected coding versus non-coding ratio of aligned reads after using Bowtie2 to remove rRNA elements in the reads. Please click here to view a larger version of this figure.
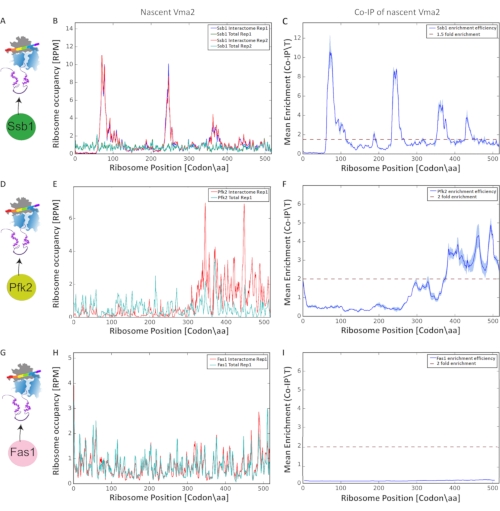
Figure 9: Co-translational interactions of three different proteins: Ssb1p, Pfk2p, and Fas1p with Vma2p, which is being synthesized by the ribosome, analyzed by SeRP. All y axes are shown in reads per million (RPM) reads.(A, D, G) Experimental scheme of SeRP of Ssb1p, Pfk2p, and Fas1p C` terminally tagged by GFP, respectively. (B, E, H) Ribosome occupancy along the orf of total translatomes compared to Ssb1, Pfk2p, and Fas1p interactomes, respectively (in biological replicates). (C, F, I) Mean enrichment of Ssb1p, Pfk2p, and Fas1p (IP/Total ratio) at each ribosome position in [codon/aa] along the orf, respectively. Variation between biological replicates is indicated by the shaded area. Please click here to view a larger version of this figure.
Table 9: 3' Linker and primer sequences. 3' Linker L1: Linker 3-L1 with 5ʹ adenylation and 3ʹ dideoxy-cytidine unique molecular identifiers ('NN…') (RNase-free HPLC purification; Reverse transcription linker: reverse transcription (L(rt)) with 5ʹ phosphorylated, unique molecular identifiers (RNase-free HPLC purification); PCR forward primer: PCRf; HPLC purified. Please click here to download this Table.
Supplementary File. Please click here to download this File.
