Classification of Neural Stem Cell Activation State In Vitro using Autofluorescence
Summary
This protocol describes strategies to identify and enrich for cell-state in primary adult mouse neural stem cell cultures by autofluorescence imaging using i) a confocal microscope, ii) a fluorescent activated cell sorter to perform intensity imaging, or iii) a multiphoton microscope to perform fluorescence lifetime imaging.
Abstract
Neural stem cells (NSCs) divide and produce newborn neurons in the adult brain through a process called adult neurogenesis. Adult NSCs are primarily quiescent, a reversible cell state where they have exited the cell cycle (G0) yet remain responsive to the environment. In the first step of adult neurogenesis, quiescent NSCs (qNSCs) receive a signal and activate, exiting quiescence and re-entering the cell cycle. Thus, understanding the regulators of NSC quiescence and quiescence exit is critical for future strategies targeting adult neurogenesis. However, our understanding of NSC quiescence is limited by technical constraints in identifying quiescent NSCs (qNSCs) and activated NSCs (aNSCs). This protocol describes a new approach to identify and enrich qNSCs and aNSCs generated in in vitro cultures by imaging NSC autofluorescence. First, this protocol describes how to use a confocal microscope to identify autofluorescent markers of qNSCs and aNSCs to classify NSC activation state using autofluorescence intensity. Second, this protocol describes how to use a fluorescent activated cell sorter (FACS) to classify NSC activation state and enrich samples for qNSCs or aNSCs using autofluorescence intensity. Third, this protocol describes how to use a multiphoton microscope to perform fluorescence lifetime imaging (FLIM) at single-cell resolution, classify NSC activation state, and track the dynamics of quiescent exit using both autofluorescence intensities and fluorescence lifetimes. Thus, this protocol provides a live-cell, label-free, single-cell resolution toolkit for studying NSC quiescence and quiescence exit.
Introduction
NSCs create newborn neurons throughout life in many organisms in a process referred to as adult neurogenesis1,2. To produce newborn neurons, a qNSC first must activate, entering the cell cycle to expand the population and produce neural progenitors3,4,5,6. Although there is much known about NSC quiescence, our ability to fully identify the drivers and regulators of NSC quiescence is constrained by technical limitations that exist to isolate and identify qNSCs and their transition to activation. Autofluorescence imaging has previously been successful in studying changes in cell state in many different cell types, such as microglia and T-cells, by resolving metabolic remodeling, which influences the optical properties of autofluorescent metabolic cofactors such as nicotinamide adenine dinucleotide phosphate (NAD(P)H) and flavin adenine dinucleotide (FAD)7,8. NSCs substantially remodel their metabolic networks as they undergo quiescence exit9,10,11,12,13,14. Thus, to take advantage of these differences, NSC autofluorescence was recently used to identify and enrich the NSC activation state by detecting shifts in autofluorescence attributed to the metabolic remodeling that occurs as NSCs exit quiescence15. Imaging autofluorescence provides several technical advantages: i) it does not require the addition of exogenous labels, which can impact cell behavior; ii) it can provide high-resolution single-cell data on the NSC activation state; and iii) it does not require the destruction of the cell7,16. This protocol outlines three strategies for harnessing NSC autofluorescence to study NSC quiescent and activated cell states15.
Recently, NSCs isolated from 6-week-old male mice from the subgranular zone of the hippocampus, cultured and reversibly put into quiescence in vitro10,13,17,18,19,20,21, were found to exhibit increased levels of punctate autofluorescence (PAF) that excite between 400-600 nm and emit between 500-700 nm. This signal was specific to qNSCs compared to activated, cycling NSCs15. The ability to visually separate these two populations without the use of additional antibody markers or reporters is useful for many experimental questions on the nature of qNSCs and quiescence exits. Thus, first, this protocol describes strategies to image the PAF in qNSCs using a confocal microscope, which can be used to identify NSC activation state. Second, this protocol describes strategies to detect the PAF using fluorescence-activated cell sorting (FACS) and further describes how to sort based on this signal to enrich qNSCs or aNSCs. These strategies provide one measure that can be used to cluster and separate NSCs based on cell state.
To develop a higher resolution method of separating NSCs not only in distinct states but also as they transition through quiescence exit towards full activation, fluorescence lifetime imaging (FLIM) was performed using a multiphoton microscope to image NAD(P)H (termed Channel 1) autofluorescence and green autofluorescence (termed Channel 2; which detects both FAD autofluorescence and PAF in qNSCs) lifetimes together with their intensity. This approach capitalizes on the fact that the optical properties of molecules in the cell are dependent on their physical properties16,22. For example, NAD(P) (NAD and NADP are optically indistinguishable, and thus NAD(P) is used to refer to both species) is not autofluorescent in the oxidized state but is autofluorescent in its reduced state (NAD(P)H)23. Further, additional physical properties of autofluorescent molecules, such as their binding status to enzymes, can be extrapolated by performing fluorescence lifetime imaging7,22,24. For example, NAD(P)H has a shorter fluorescence lifetime when not bound to an enzyme22. As autofluorescent molecules such as NAD(P)H, which is involved in hundreds of metabolic reactions, are used differently by cells progressing through different states or cell behaviors, these shifts can be detected and quantified using a multiphoton microscope detecting autofluorescence lifetime23. Together with the abundance, or intensity, of the autofluorescence, these measures provide multi-dimensional information to separate NSCs into one cell state or the other and through the dynamic transitions between states. Third, this protocol describes performing, analyzing, and interpreting FLIM and intensity measures of Channel 1 (NAD(P)H) and Channel 2 (PAF) signals using a multiphoton microscope. In summary, this protocol describes a live-cell, label-free toolkit for studying NSC quiescence that provides high-resolution single-cell data on NSC state.
Protocol
All procedures in this protocol are approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Wisconsin-Madison.
1. Using a confocal microscope to image PAF in qNSCs and aNSCs to identify NSC cell-state
- Generate qNSCs and aNSCs in vitro from primary NSC cultures.
- Culture NSCs purified from adult mouse brains in a proliferative medium for expansion (Table 1)18,20,21. Thus, by default, in vitro NSC cultures are aNSCs.
- To generate qNSCs, use the following protocol to plate aNSCs onto coated glassware and then treat them with qNSC medium to induce quiescence over 3 days.
- Prepare glassware with a refractive index suitable for oil immersion objectives for adhering NSCs by coating first with a poly-L-ornithine (PLO; 10 mg/mL in water for plastic, 50 mg/mL in water for glass) solution sufficient to cover the bottom of the dish (for a 1 cm2 imaging cuvette well, use ~150 µL) for 1 h at 37 °C in ddH2O.
- Remove the PLO solution and wash 3 times with phosphate-buffered saline (PBS).
- Coat with laminin solution (5 mg/mL) sufficient to cover the bottom of the dish (for a 1 cm2 imaging cuvette well, use ~150 µL) in PBS and incubate at minimum for 3 h at 37 °C, or overnight.
- Remove the laminin solution, then add a culture medium sufficient to cover the bottom of the dish (for a 1 cm2 imaging cuvette well, use ~150 µL).
- Prepare the cells for trypsinization by centrifuging aNSCs (they can start as monolayers or neurospheres) at 120 x g for 4 min at room temperature (RT, ~25 °C).
- Trypsinize pelleted aNSCs from the previous step in 100 µL of trypsin mix for 5 min at 37 °C and then quench the trypsinization with 200 µL of trypsin inhibitor mix (Table 1).
- Allow the NSCs to sit for 2 min, then mechanically triturate them by pipetting up and down 10 times.
- Add 5 mL of aNSC medium, and spin the cells at 120 x g for 4 min.
- Resuspend the NSCs in 1 mL of aNSC medium and plate the cells at 10%-20% confluence in aNSC medium. For example, plate ~5,000 cells into one well of a 1 cm2 imaging cuvette well with 150 µL medium.
- After allowing 24 h for aNSCs to adhere to the surface of the dish, remove the aNSC medium and replace it with qNSC medium sufficient to cover the bottom of the dish (for a 1 cm2 imaging cuvette well, use ~150 µL; Table 1– as previously described10,13,17,18) in wells in which quiescence will be induced.
- Change aNSC and qNSC medium at least once every 2 days. After 3 days in qNSC medium, NSCs are quiescent and can be used for imaging experiments.
- Use a confocal microscope to image live qNSCs and aNSCs in vitro.
NOTE: Each microscope has its proprietary software; thus, follow these steps by performing analogous actions to configure a specific microscope. This protocol will provide instructions for working in NIS-Elements.- Configure the confocal microscope for autofluorescence imaging.
- Click 405 Laser, and type in the emission window within the 405 Laser menu. Click TD to collect a transmitted light image and set the HV value to visualize the sample.
- Set the Laser Power to 3.5% and Gain to 75. Set Zoom to 2. Set up the confocal scanning parameters, set Pixel Dwell to 0.5, and set Resolution to 2048 x 2048 pixels.
- Bring the cells in focus under a ~60x oil immersion objective lens using brightfield to focus.
- Switch to confocal scanning mode by clicking Eye Port, ensuring that the light path is now going to the scan head and that no filter cube is in the light path.
- PAF is enriched in qNSCs and is widely excitable and emittable (see Figure 1). Choose the best optical configuration based on the system's capabilities using Figure 1 as a guide. For the strongest and cleanest signal, excite with a laser between ~400-500 nm, collect ~580-620 nm, and use a ~60x oil immersion objective lens.
NOTE: Autofluorescence imaging requires higher laser powers to excite than typical imaging experiments would require. If imaging other autofluorescent molecules or other labels together with PAF, carefully design the optical configuration to avoid bleed-through into autofluorescence channels. - Acquire images of autofluorescence in qNSCs and aNSCs. Click Scan, then focus on the image, and then click Capture. For the best images, collect single-plane images with the cytoplasm of cells (based on dimmer autofluorescence diffusely spread throughout the cell) in focus to get a representative cross-section of each cell.
- Use an identical optical configuration (e.g., same laser power) and image samples on the same day if aiming to directly compare different images.
- The exact powers of the laser and detector will depend on each specific system. Start by using low laser powers and then slowly increasing powers until the signal is detectable without being saturated.
- Configure the confocal microscope for autofluorescence imaging.
- Analyze PAF in live qNSCs and aNSCs in vitro.
- After acquiring autofluorescence images in qNSCs or aNSCs, quantify the images by opening a file in ImageJ25.
- Use the option Process > Filters > Gaussian Blur (Sigma: 2) on the image of qNSC/aNSC autofluorescence to smooth uneven pixels.
- Use Image > Adjust > Threshold to select pixels representing PAF (eliminate background pixels) and click on Apply. Use the same thresholding parameters for all images that will be directly compared.
- Use Process > Binary > Watershed to separate PAFs in close spatial proximity.
- Using the computer mouse, draw a region of interest (ROI) around a cell, as determined either by looking at a brightfield image or by looking at diffuse autofluorescence present evenly throughout the cytoplasm of the cell.
NOTE: It is generally fine to exclude thin peripheral processes and include the nucleus as autofluorescence is typically not present in these locations and thus would not disrupt the analysis. - Use Analyze > Analyze Particles (Size: 0.1-infinity µm; Circularity: 0-1) with summarize checked.
NOTE: ImageJ will then produce an output window with data on the particles in the region of interest, which allows for the tabulation of the number of PAF in each cell. Other fluorescent molecules can be paired with imaging PAF. However, as PAF is a relatively dim signal, it is important to validate that other fluorophores do not bleed into the PAF channel. Typically, fluorophores absorbing 600+ nm and emitting 650+ nm can be paired with imaging PAF, such as LipidSpot 610.
2. Using FACS to enrich for NSC activation state in cultured NSCs based on autofluorescence
- Generate qNSCs and aNSCs as described in section 1.
- Sort a mixture of live qNSCs and aNSCs based on autofluorescence.
NOTE: As an example, this protocol will discuss how to enrich aNSCs or qNSCs in a sample generated by mixing qNSCs and aNSCs at a 1:1 ratio.- Prior to trypsinization and cell sorting, label the cells progressing through the S-phase by adding 10 µM 5-ethynyl-2'-deoxyuridine (EdU; a thymidine analog that incorporates into cells synthesizing DNA, such as in the S-phase of the cell cycle) to the cell culture medium for 1 h.
- Trypsinize qNSCs and aNSCs as discussed in steps 1.1.8, resuspend in 0.5 mL of FACS buffer (Table 1) and then place on ice.
NOTE: qNSCs adhere strongly to the dish. At relatively higher confluence, qNSCs can be mechanically dissociated from the dish. However, if qNSCs are at a relatively lower confluence or are difficult to remove from the dish mechanically by pipetting, use trypsin to remove them from the dish.- Remove the culture medium from qNSCs and add 0.25% trypsin sufficient to cover the bottom of the dish (for a 1 cm2 imaging cuvette well, use ~150 µL), incubating for 3 min at 37 °C.
- Following incubation, quench the trypsin reaction by adding medium corresponding to each cell type equal to the volume of trypsin added. Mechanically dissociate cells from the dish before collection and centrifugation.
- Using a ~130 µm nozzle, analyze qNSCs and aNSCs using a flow cytometer or FACS with optical conditions based on the instrument's capabilities and detect PAF as shown in Figure 1. For example, use a 405 nm laser to excite the sample and use a 580-620 nm filter for detection.
- Collect at least 10,000 singlet qNSCs and aNSCs in a data file and then use these to design gates to collect an enrichment of qNSCs or aNSCs. For example, set gates to collect the top 25% or bottom 25% of NSCs based on autofluorescence intensity in the aNSC:qNSC (1:1) mixed sample.
- After setting gates to sort singlets with brighter or dimmer autofluorescence as described above, sort the cells into PLO-, laminin-coated wells filled with aNSC medium (10,000 cells/cm2, see step 1.1.3).
- Following FACS, allow NSCs to sit for 3 h in the incubator. Fix the cells by treating them with 4% paraformaldehyde sufficient to cover the bottom of the dish (for a 1 cm2 imaging cuvette well, use ~150 µL) for 15 min at RT.
NOTE: To visualize EdU in cells, it is easiest to obtain a commercial kit for detecting EdU and follow the manufacturer's recommended protocol. - First, permeabilize the cells by treatment with 0.25% triton in PBS for 15 min at RT.
- Next, treat cells with an EdU staining solution at RT for 30 min.
NOTE: The precise composition of the EdU staining solution will vary depending on the source of the reagents but roughly consists of a reaction buffer, a reaction buffer additive, copper sulfate, and an alkyne or azide-modified molecule. See the Table of Materials for a recommended kit. - Following staining to visualize EdU, wash the samples 3 times for 10 min with PBS.
3. Using a multiphoton microscope to detect Channel 1 and Channel 2 autofluorescence and perform FLIM on NSCs in vitro to identify NSC cell-state populations and transitions
NOTE: Each microscope has its proprietary software; thus, follow these steps by performing analogous actions to configure a specific microscope. This protocol will provide instructions for working in Prairie View.
- Generate qNSCs and aNSCs as described in section 1.
- Set up the multiphoton microscope for fluorescence lifetime imaging.
- Use an objective lens with 40x or greater magnification (~1.15 NA).
- Use a multiphoton microscope with the following, or equivalent, components: a tunable ultrafast laser, a 720 nm long pass dichroic, a gallium arsenide phosphide (GaAsP) photomultiplier tube (PMT), time-correlated single-photon counting electronics, and an analog power meter.
- For imaging Channel 1, set the tunable laser to 750 nm and use a 440/80 nm emission filter cube. For imaging Channel 2, set the laser to 890 nm and use a 550/100 nm emission filter cube.
NOTE: Turn off room lights before the PMT's are turned on.
- Instrument response function
- Capture an instrument response function (IRF) by imaging a crushed Urea crystal to account for the temporal response of the instrument in the measured decay.
NOTE: The IRF is convolved with the decay to accurately calculate the decay fit curve. For acquisition, the laser is set to 890nm, and the 440/80 nm emission filter cube is used. The GaAsP PMT is set at a gain of 800, and the pockels (laser power) are set very low initially, usually 1. - Select a field of view with crystalline Urea and increase the pockels slightly (up to a maximum of 15).
- Next, acquire an image using the same parameters that are used for cell imaging: constant fraction discriminator (CFD) 1 x 104 – 1 x 105, 256 x 256 resolution, dwell time set at 4.8 µs, optical zoom set at 1.5x, and a 60 s integration time.
- Capture an instrument response function (IRF) by imaging a crushed Urea crystal to account for the temporal response of the instrument in the measured decay.
- Perform fluorescence lifetime imaging and intensity imaging for Channel 1 and Channel 2 autofluorescence on live qNSCs and aNSCs in vitro
- Find a field of view to image: Use the oculars to find a suitable field of view and change the light path to the sample on the microscope to enable imaging.
NOTE: A Channel 1 image will be acquired first, followed by a Channel 2 image of the same field of view. - Setup for collection of Channel 1 image: Click on the Power/Gain tab and set the gain on the PMT to 800, click on the 2-P Laser tab and select 750 nm for the laser, and ensure that the proper filter cube is used (440/80 nm).
- Collect Channel 1 image.
- Start Vorschau mode by going to the Power/Gain tab and setting the Pockels to 30, then going to the Scanning section of that screen and clicking Live Scan.
- Increase the Pockels to find a good field of view at low laser power (below 3.6 mW). Once the field of view is found, adjust the Pockels so that the power meter reads ~3.6 mW on the power after the pockel cell, and the constant fraction discriminator (CFD) in the Count Rates tab measures 1 x 104 – 1 x 105.
- Go to the Scanning section and click Single Scan once those parameters are met. This will acquire a Channel 1 image over 60 s.
- To collect a Channel 2 image, click on the 2-P Laser tab and select 890 nm for the laser, swap the filter cube to the Channel 2 emission cube (550/100 nm).
- Start the preview mode as described in 3.4.3, increase the Pockels until the power meter reads ~7 mW, and the CFD counts are once again 1 x 104 -1 x 105.
- Go to the Scanning section and click on Single Scan once those parameters are met. This will acquire a Channel 2 image over 60 s.
- Acquire more images – After acquiring a Channel 1 and Channel 2 image, find a new field of view and repeat steps 3.4.2 – 3.4.4. Typically, collecting 4-6 fields of view to image ~50-100 cells is sufficient for one condition in an experiment.
NOTE: mCherry can be used as an additional fluorescent label without bleed-through into the Channel 1 and Channel 2 channels.
- Find a field of view to image: Use the oculars to find a suitable field of view and change the light path to the sample on the microscope to enable imaging.
- Analyze the fluorescent lifetime images.
- Generate decay matrices for fluorescence lifetime images in SPCImage following steps 3.5.2-3.1.14.
NOTE: For additional information, consult the updated handbooks freely available online26. - Open SPCImage and open the IRF FLIM image collected in section 3.3.
- Adjust the vertical black bars in the histogram to be limited to the IRF decay curve. From the top menu bar, click on IRF > Copy to Clipboard.
- Open a FLIM image file within the data set to be analyzed, obtained in section 3.4.
- From the top menu bar, click on IRF > Paste from Clipboard.
- In the bottom right of software, set Components to 2.
- In the top menu bar of the software, click on Options > Model. Then select MLE for Fit Method, Spatial Binning auf Square, Threshold auf Peak, and under Settings, select Multiexponential Decay.
- In the bottom left of the software, increase Bin until the photon count is at least 100 in representative cytoplasmic pixels at the peak of the photon count immediately after excitation.
- In the bottom left of the software, set the Threshold. For Channel 1 use a threshold of "50". For Channel 2, use a threshold of "0."
- In the top menu bar of the software (this protocol describes how to operate version 8.3), click on Calculate > Decay Matrix.
NOTE: Ensure that Chi2 values are ~0.8-1.2 throughout the image. This confirms that the data will be robust by validating biexponential decay modeling. - In the top menu bar, click on File > Save.
- In the top menu bar of the software, click on File > Export, and export the following:
Color-Coded Value, T1, T2, Chi2, Pixel Intensities, A1[%], A2[%] and Color-Coded Image (with Legend)
NOTE: Now SPCImage is set up to do a batch process, analyzing all images of a given channel (analyze Channel 1 FLIM images together and then repeat starting at step 3.5.2 for Channel 2 FLIM images). - To perform a batch process, click on Calculate > Batch Processing and select the FLIM images to be processed.
- In the top menu bar, click on File > Export > Batch Export and select the decay matrices to be exported (generated in the previous step).
- Use CellProfiler to create cytoplasmic masks following steps 3.5.16-3.5.24.
NOTE: To do this, nuclei will be manually around Channel 1 intensity images, where the nucleus is black and clearly visible. Then a CellProfiler pipeline manual_segmentation.cpproj27 (Supplementary File 1) will automatically produce a whole cell and cytoplasmic mask, as directed by the nuclear mask. Nuclei can alternatively be labeled with mCherry and imaged alongside Channel 1 and Channel 2 autofluorescence to provide the capacity to automate the identification of nuclei. Many conventional nuclear dyes, however, will spectrally bleed into the autofluorescence channels and thus are incompatible with this technique. - In R Studio, use R_ASCtoTIFF.rmd27 (Supplementary File 2) to convert the X_photons.asc files, where X is the name of the acquired image from the Channel 1 image set to TIF files.
- Open Cell Profiler and open manual_segmentation.cpproj.
- Load in the Channel 1 photon TIFs created in step 3.5.16 in the Images step at the top of the pipeline.
- In the bottom 3 steps in the pipeline titled SaveImages, set a save path for the masks CellProfiler will generate.
- Click on Start Test Mode at the bottom left of CellProfiler.
- Click on Run at the bottom left of CellProfiler. When CellProfiler gets to the IdentifyObjectsManually step, a window will appear displaying the current Channel 1 image being processed.
- Click the F key on the keyboard and then manually draw a trace of one cell's nuclei while holding down the mouse left-click button. After tracing nuclei, release the left-click button on the mouse. Repeat this step for all cells in the image.
- Click on Done in the bottom right of the pop-up window with the intensity image. The software will now finish the pipeline and export cytoplasmic, nuclear, and total cell masks.
- In the bottom left, click on Next Image Set and repeat steps 3.5.21-3.5.23 for all Channel 1 TIF images.
- Use R script (Integrate decay matrices and cytoplasmic masks.rmd) to integrate decay matrices generated in steps 3.5.2-3.5.14 and cytoplasmic masks generated in steps 3.5.15-3.5.24 to obtain average fluorescence lifetime imaging variables for each cell's cytoplasm by following steps 3.5.26-3.5.30
- Using a spreadsheet (e.g., Microsoft Excel), create a .csv document titled test_key27 [Supplementary File 3] for an example) with 3 columns.
- Title the columns as Folder, NADH, and FAD. Fill in the table to list Folder Location in Folder, and then image title prefix in NADH and FAD, respectively, to link each Channel 1 image to each Channel 2 image (see example data – Table 2).
- Generate decay matrices for fluorescence lifetime images in SPCImage following steps 3.5.2-3.1.14.
- In R Studio, open Integrate decay matrices and cytoplasmic masks.rmd27(Supplementary File 4).
- Set the following file paths as annotated in the script: Set Working Directory, Channel 1 file location, FAD file location, Mask file location, and output file location and name.
- Run the entire script. Upon successful completion of the script, an output file will be generated, including metadata and average FLIM variables.
- Perform the analysis using autofluorescence FLIM data.rmd27 (Supplementary File 5) or using any other analysis software.
Representative Results
Confocal autofluorescence imaging to separate NSC cell state (Figure 1)
To use confocal microscopy to resolve the NSC activation state, qNSCs, and aNSCs were generated in vitro using either an activation medium or quiescence medium, as described previously10,13,17,18. To detect PAF in NSCs, live qNSCs and aNSCs were imaged using the same exposure on a confocal microscope (Ex: 405 nm, Em: 580-620 nm). qNSCs exhibited a higher number of PAF compared to aNSCs (Figure 1A,B). This finding illustrates how autofluorescence properties can be used as markers to identify the cell state of qNSCs and aNSCs.
FACS enrichment of NSC cell state using autofluorescence (Figure 2)
To enrich for cell cycle state using FACS, qNSCs and aNSCs were generated in vitro10,13,17,18,19 as described in this protocol and pre-labeled with EdU for 1 h prior to trypsinization and analysis in the flow cytometer to label cells progressing through S-phase. qNSCs and aNSCs were then analyzed by flow cytometry either separately or mixed at a ratio of 1 qNSC:1 aNSC (Figure 2). Gates were drawn to enrich for aNSCs or qNSCs from the mixed population, and cells were then sorted based on these gates. After FACS, cells in each sample were plated onto PLO-, laminin-coated glassware and allowed to adhere to the dish for 3 h before being fixed, stained, and analyzed for %EdU+ cells. Expectedly, cells sorted from the high autofluorescence gate were less EdU+ than the Mix sample, and samples sorted through the low autofluorescence gate were more proliferative than the Mix sample (Figure 2C). This finding confirms the capacity to enrich for NSC activation state from a heterogeneous mixture of qNSCs and aNSCs using FACS.
Multiphoton fluorescence lifetime imaging to classify NSC cell state (Figure 3)
qNSCs and aNSCs were generated in vitro and then imaged using a multiphoton microscope to perform FLIM on Channel 1 autofluorescence (Ex: 750 nm (2P), Em: 360-520 nm) and Channel 2 (Ex: 890 nm (2P), Em: 450-650 nm) (Figure 3A, Table 2). qNSCs and aNSCs exhibited autofluorescent profiles that were largely significantly different. For example, qNSCs had a higher Channel 1 fluorescence mean lifetime τm, but a lower α1 compared with aNSCs. To evaluate the capacity of NSC FLIM autofluorescence data to predict NSC activation state, a logistic regression model was generated with Channel 1 intensity, α1, τ1, τ2 and Channel 2 intensity, α1, τ1, τ2. A receiver operator curve illustrates that these data are sufficient to create a near-perfect model (Area under the curve = 0.963), accurately predicting the NSC activation state. Together, these data illustrate the capacity of FLIM and autofluorescence to be used to classify the NSC cell state.
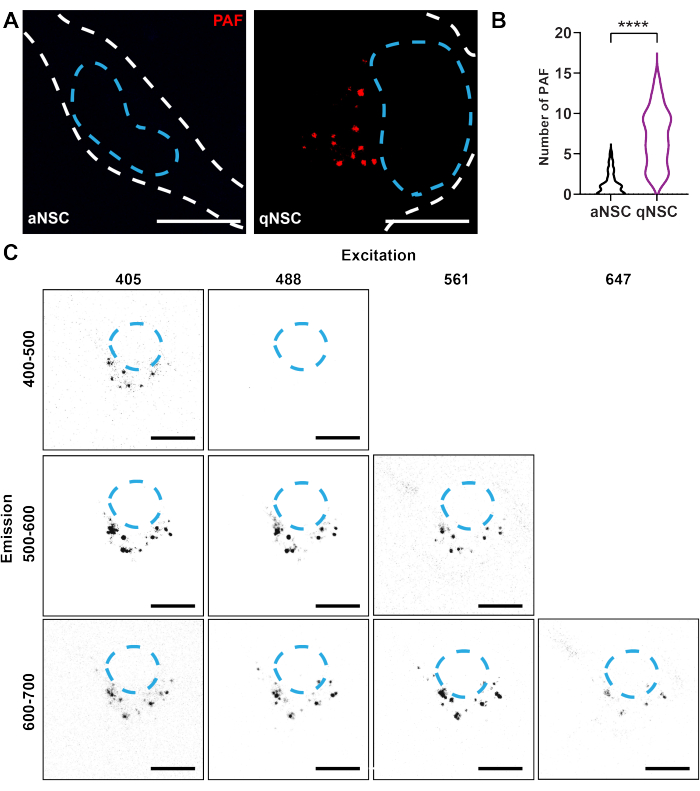
Figure 1: Confocal imaging resolves autofluorescent biomarkers of NSC activation state. (A–B) qNSCs (purple) and aNSCs (black) were imaged using the same exposure on a confocal microscope (red; Ex. 405 nm, Em 580-620 nm) and analyzed for the number of PAF (N = 3, Mann-Whitney test, mean ± SD). White dashed lines denote the edge of the cell, and blue dashed lines denote nuclei. (C) Autofluorescence in the same qNSC was imaged using various excitation and emission conditions, as indicated in the figure, with identical laser power and gain. Scale bars: 10 µm. **** p < 0.0001. Please click here to view a larger version of this figure.
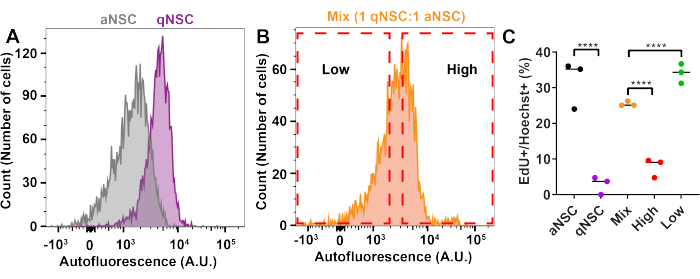
Figure 2: FACS can enrich for NSC activation state. (A–C) qNSCs, aNSCs or a qNSCs: aNSCs mix (1:1) were treated with EdU for 1 h, trypsinized, and then analyzed by flow cytometry (Ex: 405 nm, Em: 580-620 nm). Mix cells (1:1) were then sorted by FACS for cells that had either low or high autofluorescence, plated, and analyzed for proliferation by measuring the percentage of cells that were EdU+. Arbitrary units is abbreviated as "A.U." (N = 4, two-way ANOVA with post hoc Tukey's test, mean ± SD). **** p < 0.0001. Please click here to view a larger version of this figure.
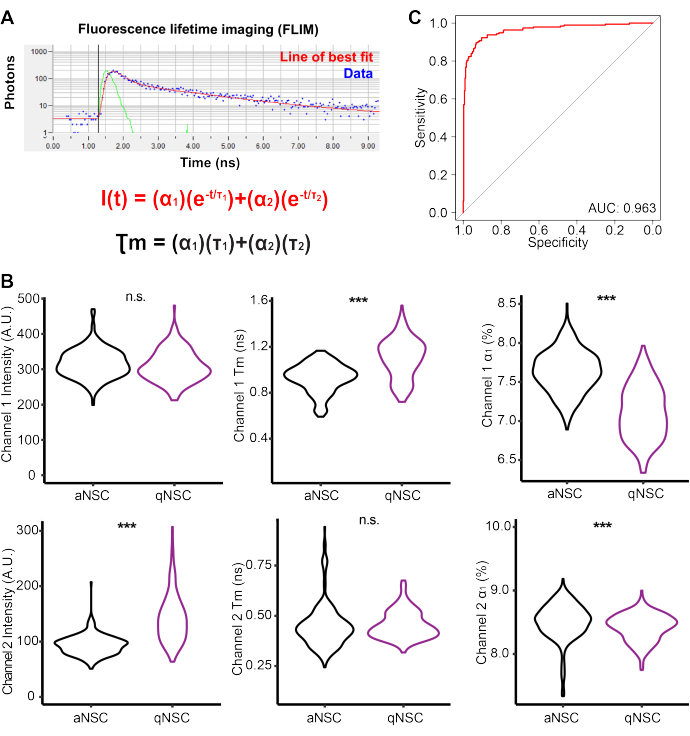
Figure 3: Multiphoton fluorescence lifetime imaging reveals biomarkers of NSC activation state. (A) Schematic depicting curve fitting analysis of acquired FLIM data. (B) Channel 1 and Channel 2 FLIM measurements, including the intensity, mean lifetime (Tm), and fractional contribution (α1) values for qNSC (purple) and aNSC (black) data (n = 501 cells, two-sided logistic regression, generalized linear model). (C) Receiver operator curve demonstrating a logistic regression model generated using Channel 1 intensity, α1, τ1, τ2, and Channel 2 intensity, α1, τ1, τ2 to classify NSCs as aNSCs or qNSCs. *** p < 0.001. Please click here to view a larger version of this figure.
Table 1: Media and solutions. Recipes for all solutions used in this protocol. Please click here to download this Table.
Table 2: Example data. Representative qNSC and aNSC FLIM data for Channel 1 and Channel 2 autofluorescence. Please click here to download this Table.
Supplementary File 1: manual_segmentation.cpproj Please click here to download this File.
Supplementary File 2: R_ASCtoTIFF.rmd Please click here to download this File.
Supplementary File 3: test_key Please click here to download this File.
Supplementary File 4: Integrate decay matrices and cytoplasmic masks.rmd Please click here to download this File.
Supplementary File 5: autofluorescence FLIM data.rmd Please click here to download this File.
Discussion
This protocol describes a live-cell, label-free, non-destructive, single-cell resolution technique that allows for the classification of NSC cell-state in vitro through imaging of autofluorescent signals in NSCs. This approach detects metabolic shifts that occur during NSC quiescence exit, which influence the optical properties of metabolic cofactors, such as NAD(P)H, and offers many advantages over existing technologies to study NSC quiescence. For example, many conventional techniques for studying qNSCs and aNSCs, such as labeling with cell cycle markers like EdU, require the fixation of samples. Methods that currently exist that are capable of studying live NSCs are further limited by requiring the introduction of exogenous labels, typically by generating fluorescent protein-encoding transgenes. These tools are limited both in the resources required to generate them and many technical caveats. For example, the intermediate filament proteins nestin and Glial Fibrillary Acidic Protein (GFAP) are commonly used as markers of qNSCs and aNSCs12,13,18,28,29. However, nestin and GFAP are known to be differentially expressed between qNSCs and aNSCs12,13,18,28,29. Further, autofluorescence imaging provides high-resolution single-cell data, which can unravel single-cell heterogeneity that is lost or complicated to interpret in experiments that culminate in a bulk analysis of a population of cells.
However, autofluorescence imaging also has several limitations. Many of the autofluorescent signals become lost upon cell fixation. Thus, autofluorescence imaging is largely limited to the study of living cells. Autofluorescence imaging also relies upon a lack of exogenously introduced fluorophores, which can bleed into autofluorescent channels. Although many fluorophores can be compatible with autofluorescence imaging in specific situations, pairing autofluorescence imaging with many existing tools is not always possible. Live-cell imaging can also induce phototoxicity. One iteration of imaging using this protocol does not induce sufficient phototoxicity to reduce NSC viability. However, repeated imaging of cells in a time course at more frequent intervals many times per day may generate significant phototoxicity and, thus, is not a viable strategy for studying NSC quiescence. Lastly, although autofluorescence imaging can be used to study NSCs from 6-week-old male mice from either the hippocampus or the lateral ventricles, it is unclear how broadly this technique may be used to study NSCs from other sources, ages, or other stem cell types.
The protocol described here also assumes accurate production of qNSCs and aNSCs in vitro using previously established protocols10,13,17,18,19. If reproducing the results of this protocol is challenging, ensure that qNSCs and aNSCs are being properly generated by probing for the presence or absence of various markers of qNSCs and aNSCs, as previously described10,13,17,18,19. Taken together, autofluorescence imaging provides a novel technical approach to identifying qNSCs and aNSCs and studying NSC quiescence exits.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
We thank the UW-Madison flow cytometry core (P30 CA014520 and 1S10RR025483-01), and members of the Moore lab and UW-Madison community for their input. We thank our funding sources: NIH T32 T32GM008688 (to C.S.M.), Diana Jacobs Kalman Fellowship from AFAR (to C.S.M.), Wisconsin Graduate Fellowship (to C.S.M.), DP2 NIH New Innovator Award (1DP2OD025783, to D.L.M.), Vallee Scholar Award (to D.L.M.), NIH 1R56NS130450 (to D.L.M and M.C.S.), R01 CA185747 (to M.C.S.), R01 CA205101 (to M.C.S.), R01 CA211082 (to M.C.S.), and the National Science Foundation Grant No. CBET-1642287 (to M.C.S.).
Materials
| 40x Water objective lens | Nikon | MRD77410 | Objective lens used in multiphoton microscope in Part 3 |
| 8 well cuvette | Ibidi | 80826-90 | For imaging aNSCs/qNSCs |
| Analog power meter | Thorlabs | PM100A | Used in multiphoton microscope in Part 3 |
| Antibiotic-Antimycotic (100X) (PSF) | Thermo Fisher | 15240062 | Antibiotic for NSC media |
| B-27 | Invitrogen | 17504044 | Nutrient supplement for NSC media |
| BMP4 | Fisher Scientific | 5020BP010 | Factor for inducing quiescence |
| Bovine serum albumin | Sigma | A4919-25G | For making BMP4 |
| Chameleon ultrafast laser | Coherent | N/A | Laser used in multiphoton microscope in Part 3 |
| Confocal microscope | Nikon | C2 | Microscope used for Part 1 |
| DMEM/F-12 (without GlutaMAX) | Invitrogen | 11320033 | Base media for NSCs |
| DNAse | Sigma | D5025-15KU | Added to trypsin inhibitor |
| EdU assay kit | Invitrogen | C10337 | Proliferation assay for cell culture |
| EGF | PeproTech | AF-100-15-500UG | Growth factor for NSC media |
| FGF | PeproTech | 100-18B | Growth factor for NSC media |
| Fluorescent activated cell sorter | BD | FACSAria | Fluorescent Activated Cell Sorter used for Part 2 |
| Heparin | Sigma | H3149-50KU | Additive for NSC media |
| L-15 | Invitrogen | 21083027 | For preparing trypsin inhibitor solution |
| Laminin | Sigma | L2020-1MG | For coating glassware |
| Nikon TiE inverted microscope | Nikon | N/A | Microscope frame Used for Part 3 |
| PLO | Sigma | P3655-100MG | For coating glassware |
| SPC-150 Single photon counting electronics | Becker and Hickl | N/A | Used in multiphoton microscope in Part 3 |
| Trypsin (for trypsinizing pellets of aNSCs that were growing as spheres or monolayers) | Gibco | 15090046 | For trypsinizing neurospheres or adherent aNSCs |
| Trypsin (for trypsinizing qNSCs) | Gibco | 25200072 | For trypsinizing adherent qNSCs |
| Trypsin inhibitor | Sigma | T6522-100MG | For inhibiting trypsinization of aNSCs |
| Urea crystals | Sigma | U5128-5G | Used to collect an IRF |
| Versene | Thermo Fisher | 15040066 | For preparing trypsin |
Referenzen
- Goncalves, J. T., Schafer, S. T., Gage, F. H. Adult neurogenesis in the hippocampus: From stem cells to behavior. Cell. 167 (4), 897-914 (2016).
- Kuhn, H. G., Dickinson-Anson, H., Gage, F. H. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 16 (6), 2027-2033 (1996).
- Silva-Vargas, V., Maldonado-Soto, A. R., Mizrak, D., Codega, P., Doetsch, F. Age-dependent niche signals from the choroid plexus regulate adult neural stem cells. Cell Stem Cell. 19 (5), 643-652 (2016).
- Urban, N., Blomfield, I. M., Guillemot, F. Quiescence of adult mammalian neural stem cells: A highly regulated rest. Neuron. 104 (5), 834-848 (2019).
- Urban, N., et al. Return to quiescence of mouse neural stem cells by degradation of a proactivation protein. Science. 353 (6296), 292-295 (2016).
- Kalamakis, G., et al. Quiescence modulates stem cell maintenance and regenerative capacity in the aging brain. Cell. 176 (6), 1407-1419 (2019).
- Walsh, A. J., et al. Classification of T-cell activation via autofluorescence lifetime imaging. Nat Biomed Eng. 5 (1), 77-88 (2021).
- Sagar, M. A. K., et al. Microglia activation visualization via fluorescence lifetime imaging microscopy of intrinsically fluorescent metabolic cofactors. Neurophotonics. 7 (3), 035003 (2020).
- Knobloch, M., et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 493 (7431), 226-230 (2013).
- Knobloch, M., et al. A fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 20 (9), 2144-2155 (2017).
- Knobloch, M., et al. SPOT14-positive neural stem/progenitor cells in the hippocampus respond dynamically to neurogenic regulators. Stem Cell Reports. 3 (5), 735-742 (2014).
- Shin, J., et al. Single-cell RNA-Seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 17 (3), 360-372 (2015).
- Leeman, D. S., et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 359 (6381), 1277-1283 (2018).
- Beckervordersandforth, R. Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 3 (1), 73-87 (2017).
- Morrow, C. S., et al. Autofluorescence is a biomarker of neural stem cell activation state. Cell Stem Cell. , (2024).
- Kolenc, O. I., Quinn, K. P. Evaluating cell metabolism through autofluorescence imaging of NAD(P)H and FAD. Antioxid Redox Signal. 30 (6), 875-889 (2019).
- Mira, H., et al. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell. 7 (1), 78-89 (2010).
- Morrow, C. S., et al. Vimentin coordinates protein turnover at the aggresome during neural stem cell quiescence exit. Cell Stem Cell. 26 (4), 558-568 (2020).
- Martynoga, B., et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 27 (16), 1769-1786 (2013).
- Bin Imtiaz, M. K., Jessberger, S. Isolation of adult mouse hippocampal neural stem cells for fluorescence loss in photobleaching assays. STAR Protoc. 2 (3), 100695 (2021).
- Guo, W., Patzlaff, N. E., Jobe, E. M., Zhao, X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 7 (11), 2005-2012 (2012).
- Lakowicz, J. R., Szmacinski, H., Nowaczyk, K., Johnson, M. L. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci U S A. 89 (4), 1271-1275 (1992).
- Blacker, T. S., Duchen, M. R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med. 100, 53-65 (2016).
- Datta, R., Heaster, T. M., Sharick, J. T., Gillette, A. A., Skala, M. C. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J Biomed Opt. 25 (7), 1-43 (2020).
- . ImageJ Available from: https://imagej.nih.gov/ij/ (2021)
- . Becker & Hichl Handbooks Available from: https://imagej.nih.gov/ij/ (2024)
- . GitHub Materials Available from: https://github.com/chrismorrow5/Neural-Stem-Cell-Autofluorescence-Analysis/ (2024)
- Codega, P., et al. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. 82 (3), 545-559 (2014).
- Llorens-Bobadilla, E., et al. Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell. 17 (3), 329-340 (2015).

.