Förster resonance energy transfer (FRET) has been a major driver in molecular biological and biophysical research, as it allows the investigation of processes at sub-nanometer resolution. As the efficiency of energy transfer between donor and acceptor fluorophores strongly depends on the inter-dye distance in the sub-nanometer to nanometer range, it has been effectively used as a spectroscopic ruler to explore static and dynamic conformation of biomolecules1,2,3,4. Additionally, the FRET phenomenon has been widely used for colocalization studies of membrane-associated and intracellular proteins on a bulk level5,6. In the last two decades, the method was adapted for monitoring smFRET events7, which helped to substantially increase temporal and spatial resolution and resolved even rare subpopulations in heterogeneous samples. Equipped with these techniques, unique insights were gained into the dynamics of molecular machinery such as the transcript processing rate of RNA polymerase II8, replication speed of DNA polymerases9,10, nucleosome translocation rate11, transcript splicing and stalling rate of assembled spliceosomes12, the activity of ribosomal subpopulations13, and the walking speed of kinesin motors14, to name a few. Receptor-ligand interaction durations15 and molecular forces16 have been quantified.
Intensity-based smFRET studies typically rely on sensitized emission to measure FRET efficiency: a beam splitter in the emission path spatially separates light originating from donor and acceptor fluorophores upon donor excitation, allowing for the quantification of individual fluorescence intensities. The efficiency can subsequently be calculated as the fraction of photons emitted by the acceptor with respect to the total photon count17. In addition, acceptor excitation following donor excitation (ALEX) permits measurement of the stoichiometry of the FRET events, aiding in the discrimination between true low FRET signals from signals arising, e.g., from probes featuring a photobleached acceptor fluorophore18.
Single-molecule FRET experiments are commonly carried out in one of two ways. First, a small region in the sample volume is illuminated using a confocal microscope. Single probe molecules in solution are excited when they happen to diffuse within the focal volume. With this technique, fast photon-counting detectors can be used, enabling sub-microsecond time resolution. Second, probes are specifically immobilized on surfaces and monitored via widefield microscopy, often using total internal reflection (TIR) configuration to minimize background fluorescence. Probe immobilization allows for much longer recording times than using the first approach. In addition, the larger field of view permits the monitoring of multiple probes in parallel. The need for a camera makes this method slow compared to the one described above. Time resolution is limited to the millisecond to second range.
If long time traces are required, e.g., for studying dynamic processes on a millisecond to second time scale, the first method is not applicable, as the fluorescence bursts are typically too short. The second approach fails whenever immobilization is not feasible, e.g., in live-cell experiments featuring probes diffusing within the cell membrane. Furthermore, it has been observed that biological model systems can vary their response dramatically depending on the mobility of the contacted surface16.
While combined smFRET and single-particle tracking experiments recording mobile FRET probes have been performed in the past19, there is no publicly available software for the evaluation of the data. This prompted the development of a new analysis platform, which allows for the determination of multiple properties of mobile fluorescent probes, including smFRET efficiency and stoichiometry, positions with sub-pixel accuracy, and fluorescence intensities as functions of time. Methods for filtering the resulting traces by examining stepwise bleaching behavior, nearest-neighbor distances, emission intensities, and other traits were established to exclusively choose correctly synthesized and functional single-probe molecules. The software also supports experimental and analytical techniques recently agreed upon in a multilaboratory study to produce reliable, quantitative smFRET data17. In particular, the implementation adheres to the validated procedures for the calculation of FRET efficiency and stoichiometry. Fluorescence intensities upon donor excitation in the donor emission channel IDD and acceptor emission channel IDA are used for the calculation of the apparent FRET efficiency Eapp using Eq (1).
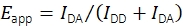 (1)
(1)
With the help of the fluorescence intensity in the acceptor emission channel upon acceptor excitation IAA, the apparent stoichiometry is calculated using Eq (2).
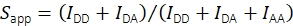 (2)
(2)
The FRET efficiency E and the stoichiometry S can be derived from Eapp and Sapp by considering four correction factors.
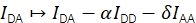
α describes the leakage of donor fluorescence into the acceptor emission channel and can be determined using a sample containing only donor fluorophores or by analyzing parts of trajectories where the acceptor has been bleached. δ corrects for the direct excitation of the acceptor by the donor excitation light source and can be measured using a sample with only acceptor fluorophores or by analyzing parts of trajectories where the donor has been bleached.
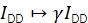 .
.
γ scales IDD to rectify diverging detection efficiencies in donor and acceptor emission channels and different quantum efficiencies of the fluorophores. The factor can be computed by analyzing the increase in donor intensity upon acceptor bleaching in trajectories with high FRET efficiencies20 or by studying a sample featuring multiple discrete FRET states.
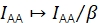
β scales IAA to correct for disparate efficiencies of donor and acceptor excitation. If γ was determined via acceptor bleaching analysis, β could be calculated from a sample of known donor-to-acceptor ratio21. Otherwise, the multi-state FRET sample also yields β.
Together, the corrections allow the calculation of the corrected FRET efficiency using Eq (3).
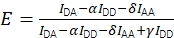 (3)
(3)
and the corrected stoichiometry using Eq (4).
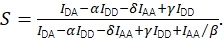 (4)
(4)
Ideally, the corrected stoichiometry for a 1:1 donor-to-acceptor ratio gives S = 0.5. In practice, a reduced signal-to-noise ratio produces a spread of the measured values of S, hampering the discrimination from donor-only signals (S = 1) and acceptor-only signals (S = 0). The resulting time traces can be used as input for a more detailed analysis of the single-molecule trajectories to obtain information such as spatiotemporal force profiles16, the mobility of the single-molecule events22, or transition kinetics between different states1.
The following protocol describes experimental parameters and procedures for smFRET tracking experiments, as well as the working principle behind data analysis using the newly developed software suite. For the acquisition of experimental data, it is recommended to use a microscopy setup meeting the following requirements: i) capability of detecting the emission of single dye molecules; ii) widefield illumination: in particular for live-cell experiments, total internal reflection (TIR23,24,25) configuration is recommended; iii) spatial separation of emission light according to wavelength such that donor and acceptor fluorescence is projected onto different regions of the same camera chip25 or different cameras; iv) modulation of light sources for donor and acceptor excitation with millisecond precision, e.g., using directly modulatable lasers or modulation via acousto-optic modulators. This permits stroboscopic illumination to minimize photobleaching of fluorophores as well as alternating excitation to determine stoichiometries; v) output of one file per recorded image sequence in a format that can be read by the PIMS Python package26. In particular, multipage TIFF files are supported.
1. Software prerequisites
- Install the miniconda Python distribution27 (minimum required Python version: 3.7).
- Open an Anaconda prompt in the Windows Start menu, or open a terminal and execute conda activate if using Linux or macOS.
- Enable the community-maintained conda-forge package repository28 by executing the following commands:
conda config –add channels conda-forge
conda config –set channel_priority strict
conda update –all - Install the required Python packages by executing:
conda install opencv trackpy lmfit ipympl scikit-learn pyqt sdt-python jupyterlab - Become familiar with JupyterLab, the user interface of the analysis software (refer to the software documentation29).
- Install the git version control system, which will be used later to download and update the analysis software. If using Linux, use the distribution's package management software to download and update. Otherwise, execute:
conda install git - Optionally install the sidecar Python package to display datasets after filtering steps during analysis:
conda install sidecar
2. Measurement of samples
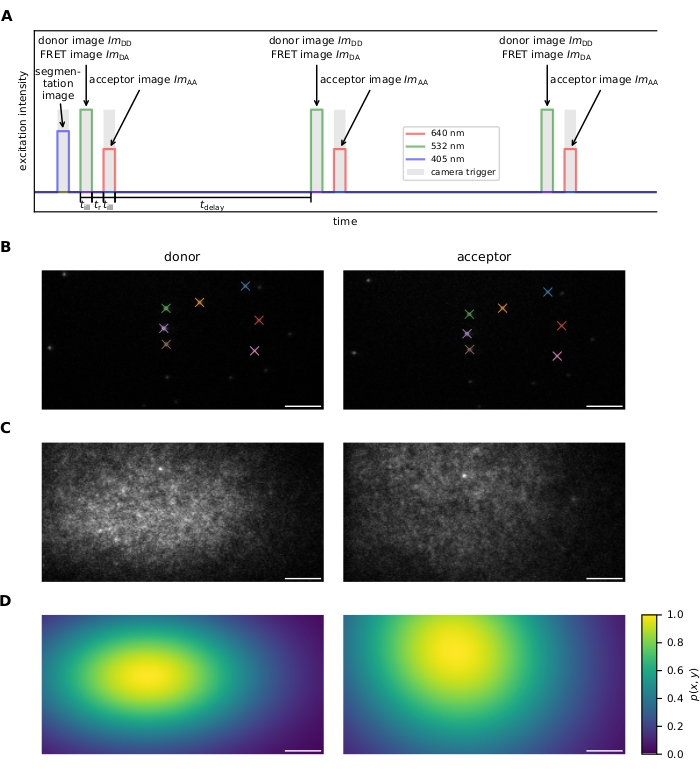
Figure 1: Image Acquisition. (A) Excitation sequence. After recording an optional image of a dye-loaded cell using the 405 nm laser, donor and acceptor are excited alternately and repeatedly for illumination time till using 532 nm and 640 nm lasers, respectively. The time tr between donor and acceptor excitation must be long enough to allow for image readout by the camera. The delay time tdelay can be used to adjust the acquisition frame rate and, therefore, the observation time span before photobleaching. This panel is modified from 16. (B) Fiducial markers are used for the calculation of the coordinate transforms between the two emission channels. Matching fiducials are indicated by color. Several shifted images should be recorded to ensure that the whole field of view is covered. (C) Laser profiles for flatfield correction are recorded using a densely labeled sample. The acceptor profile is recorded and photobleached, followed by acquisition of the donor profile. Multiple images should be taken at different sample regions, averaged, and smoothed to mitigate the influence of sample imperfections (e.g., the bright spot in the center-top of the image). (D) Flatfield correction map p(x,y) calculated from 20 laser profile recorded as described in C. Abbreviations: FRET = Förster resonance energy transfer; ImDD = donor emission image upon donor excitation; ImDA = acceptor emission image upon donor excitation; ImAA = acceptor emission image upon donor excitation. Scale bars = 5 µm. Please click here to view a larger version of this figure.
- When using an electron-multiplying charge-coupled device (EMCCD) camera, enable the EM gain to observe single-molecule signals at high signal-to-noise ratios (refer to the manufacturer's instructions).
- Excitation sequence (see Figure 1A for further details).
- Optionally record an image for segmentation to restrict data analysis to certain regions in the field of view. For instance, excite Fura-2-loaded cells using a 405 nm laser and capture their emission around 510 nm to evaluate only probes located in interfaces between cells and supported lipid bilayers (SLBs). Consequently, wait for time tr to permit camera readout.
NOTE: On EMCCD cameras, tr depends on the number of lines in the chosen region of interest (ROI). Therefore, choosing a small ROI can be advantageous because it reduces the delay between frames and the size of recorded data. Additionally, enabling frame transfer mode allows for further reduction of tr. - Alternately excite donor and acceptor fluorophores repeatedly.
- Excite the donor for an illumination time till (5-10 ms is typically short enough to avoid motion blur) while also triggering the camera.
- Wait for time tr to permit camera readout.
- Excite the acceptor for while triggering the camera.
- Wait for a time tdelay.
NOTE: This must be longer than tr to enable readout by the camera but can otherwise be chosen arbitrarily. It shall balance the requirements for time resolution and trace length. - Repeat steps 2.2.2.1-2.2.2.4. Choose the number of repeats to be large enough to ensure photobleaching of at least one fluorophore per probe within the field of view, which permits stepwise photobleaching analysis for discrimination of single-molecule signals from aggregates.
NOTE: Choosing appropriate till and excitation laser intensities commonly requires some experimentation: The longer the illumination times and the higher the laser intensities, the better is the signal-to-noise ratio in the resulting images, but the shorter are the resulting time traces.
- Optionally record an image for segmentation to restrict data analysis to certain regions in the field of view. For instance, excite Fura-2-loaded cells using a 405 nm laser and capture their emission around 510 nm to evaluate only probes located in interfaces between cells and supported lipid bilayers (SLBs). Consequently, wait for time tr to permit camera readout.
- Record a sufficient number of movies for each sample.
3. Additional measurements for the determination of correction factors
- Record a series of randomly placed fiducial markers visible in both emission channels for image registration (i.e., finding the transformation that maps coordinates of the donor emission channel onto the acceptor emission channel and vice versa). See Figure 1B.
NOTE: Image registration is performed by the software; see step 6.1.4. - Measure the intensity profile for both donor and acceptor excitation light sources for flatfield correction (i.e., correcting for inhomogeneous excitation across the field of view). To this end, prepare a sample featuring a high density of FRET probes and first acquire an image upon acceptor excitation, followed by photobleaching of the acceptor and subsequent recording of an image upon donor excitation. For increased stability, repeat several times in different sample regions. See Figure 1C,D. Alternatively, record a sample decorated with only the donor molecule and a second sample decorated with only the acceptor fluorophores.
NOTE: Flatfield correction is performed by the analysis software; see step 8.1.2. - Record a single-molecule sample (as in section 2) of a probe without an acceptor fluorophore to determine donor emission leaking into the acceptor channel.
NOTE: Donor leakage can also be computed from the actual probes' time traces after acceptor bleaching. If a sufficient number of such events is recorded, no additional measurement is necessary. Both options are supported by the analysis software; see Supplemental Information, section 3.15. - Acquire recordings of a probe without a donor fluorophore for the quantification of direct acceptor excitation by the donor excitation light source.
NOTE: Direct acceptor excitation can also be derived from the actual probes' time traces after donor bleaching. If a sufficient number of such events is recorded, no additional measurement is necessary. Both options are supported by the analysis software; see Supplemental Information, section 3.15. - Record a single-molecule sample featuring two distinct FRET efficiencies to correct for differing detection efficiencies of the donor and acceptor emission channels and differing quantum yields of the dyes.
NOTE: Such samples could be, for instance, Holliday junctions1, which fluctuate between two conformations, or DNA rods that have FRET pairs attached at different, well-defined distances. If probes feature high and sufficiently constant FRET efficiencies, the correction can also be computed from acceptor bleaching events of probes' time traces, in which case no additional measurements are necessary. Both options are supported by the analysis software; see Supplemental Information, section 3.15.
4. Single-molecule localization algorithms
NOTE: Several analysis steps require single-molecule localization. Choose between a Gaussian fitting algorithm30 and center-of-mass computation31, depending on signal density, background, and signal-to-noise ratio.
- To perform Gaussian fitting, choose the 3D-DAOSTORM30 algorithm via the respective user interfaces.
NOTE: 3D-DAOSTORM is designed to distinguish even signals with overlapping point spread functions. While this is generally an advantage, it comes with a caveat: single, bright signals are occasionally identified as two adjacent ones, which can confuse the tracking algorithm and result in the detection of two short trajectories instead of a single long one.
Set the following parameters (for details, see the documentation of the sdt-python library32, which provides the algorithm's implementation).- radius: Set the initial σ value of the Gaussian fit function in pixels depending on the effective pixel size.
- threshold: Set a minimum amplitude (i.e., brightest pixel value, corrected for the estimated local background) for a local intensity maximum to be fit.
NOTE: The threshold is arguably the most important parameter. If set too low, noise may be considered a fluorescence signal, and bright signals may be fitted with two Gaussians. If set too high, dim signals are not fit. - model: Set to 2d to fit circular Gaussians.
NOTE: The other models are not applicable to the smFRET data. - find filter: Apply a filter before finding local maxima to reduce noise, which is helpful in low signal-to-noise ratio situations. This can be i) identity: no filter; ii) Crocker-Grier: bandpass filter from Crocker-Grier algorithm31,33; or iii) Gaussian: a gaussian blur with σ set by the sigma parameter.
NOTE: For Crocker-Grier, the feat. size parameter should be roughly the radius of a point spread function in pixels.
NOTE: Fitting is performed using unfiltered raw data. - min. distance: Fit two prospective signals separated by fewer than min. distance pixels by a single Gaussian.
NOTE: This can help in the aforementioned scenario where a bright signal is wrongly detected as two adjacent signals. - size range: Select minimum and maximum σ of the fits to remove detections from spurious signals due to noise.
- Choose the Crocker-Grier algorithm via the respective user interfaces to perform center-of-mass computation (a refined algorithm31 based on Crocker's and Grier's idea33).
NOTE: This algorithm is very robust even in low signal-to-noise scenarios and in dealing with signals featuring a range of intensities but cannot fit molecules with overlapping point spread functions precisely.- radius: Set the radius (in pixels) of a disk large enough to contain the whole point spread function.
- signal thresh.: Set the minimum amplitude (brightest pixel above estimated background) for a local intensity maximum to be analyzed.
NOTE: If set too low, noise may be considered a fluorescence signal. If set too high, dim signals are not fit. - mass thresh.: Set the minimum total intensity (sum of background-corrected pixel values) of a signal to be analyzed.
NOTE: Same considerations as above apply.
5. Software initialization
- Download analysis scripts. In an Anaconda prompt, navigate to a folder to save the analysis (using the cd command) and execute
git clone https://github.com/schuetzgroup/fret-analysis.git target folder- Replace the target folder with a descriptive name such as 2021-06-14_Force-FRET-experiment.
NOTE: The analysis software will end up in this folder; ensure that this folder does not exist beforehand. It is recommended to download a copy of the analysis scripts for each experiment. This way, it is possible to revisit the analysis later, recall the parameters used, and make changes.
- Replace the target folder with a descriptive name such as 2021-06-14_Force-FRET-experiment.
- Copy Jupyter notebooks (01. Tracking.ipynb, 02. Analysis.ipynb, 03. Plots.ipynb) into the newly created folder (henceforth referred to as the root folder). If this is the first time using the software, get them from the notebooks subfolder of the root folder.
NOTE: If similar datasets have already been analyzed, copying the notebooks from a previous experiment can be a convenient option, as parameters may have changed only slightly. - Start the JupyterLab server by executing the following command in the Anaconda prompt to open a web browser window displaying JupyterLab.
jupyter lab
NOTE: The browser is only the interface, while the process running in the Anaconda prompt is doing the actual work. As a consequence, closing the browser window has only minimal effect; the session can be restored by accessing http://localhost:8888. However, interrupting the JupyterLab process in the prompt or closing the prompt will terminate the analysis, leading to the loss of unsaved work. - In the JupyterLab browser window, use the left pane to navigate to the root folder. Double-click on 01. Tracking.ipynb to launch the first notebook. After launching, look for a new tab to appear, which displays boxes, so-called cells, of Python code.
NOTE: All Jupyter notebooks feature comments describing the functionality of each code cell. Additionally, documentation for every method call can be displayed by placing the text cursor immediately before the opening ( and press Shift+Tab. - See Figure 2 for an overview of the data analysis process.
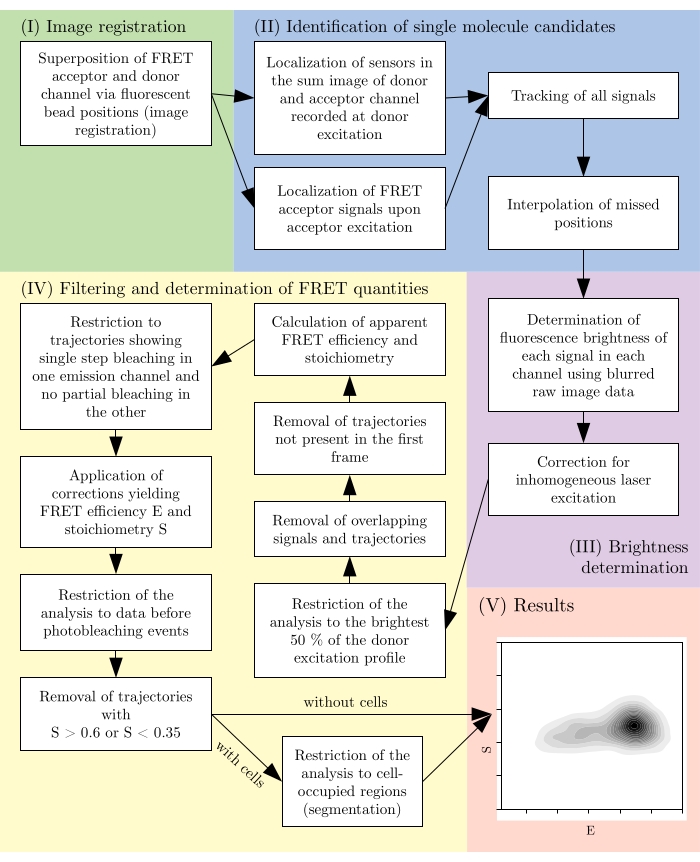
Figure 2: Overview of a typical analysis pipeline. Note that filtering steps are subject to adaptation according to the experimental design. This figure is modified from 16. Abbreviation: FRET = Förster resonance energy transfer. Please click here to view a larger version of this figure.
NOTE: Sample data to try out the software can be downloaded from https://github.com/schuetzgroup/fret-analysis/releases/tag/example_files
6. Localization, tracking, and fluorescence intensity analysis of single molecules (01. Tracking.ipynb).
- Use the 01. Tracking.ipynb Jupyter notebook for the reliable analysis of fluorescence intensity values of single-molecule signals, which is tailored for the precise quantification, particularly of faint signals often occurring in FRET measurements (e.g., due to low donor signals at high FRET events and vice versa).
NOTE: To this end, direct integration of the pixel intensities in the raw data with correction for the local background is implemented. For screenshots of each analysis step and description of function call parameters, refer to the Supplemental Information.- Specify the illumination sequence to permit the selection of donor and acceptor excitation frames, as well as frames for image segmentation from recorded image sequences.
NOTE: As the software allows the processing of data recorded with arbitrary illumination protocols, it is necessary to indicate which frame in an image sequence was acquired while exciting what type of fluorophore; see Supplemental Information, section 1.2, step 3. The frame numbers of the original image sequence are preserved. - Describe and load datasets. Analyze multiple datasets at once, provided they were recorded using the same illumination settings. Assign an identifier and a pattern that matches the respective image sequence file names to each dataset. Additionally, define specific datasets for special purposes, such as recordings of fiducial markers for image registration, excitation light profiles for flatfield correction, and optionally donor-only and acceptor-only samples to determine correction factors.
- Select emission channels in raw images if both channels were recorded using a single camera. For this, use the appropriate graphical widget to select the appropriate regions for donor and acceptor emission.
- Localize fiducial markers in both emission channels and perform image registration. Use the provided user interface to find the appropriate parameters for the localization algorithm for both the donor and the acceptor emission channels. See section 4 for information about supported localization algorithms.
NOTE: Randomly distributed fiducial markers can be identified across emission channels by the spatial distribution of their nearest neighbors (Figure 1B). A custom implementation of the algorithm proposed for selective plane illumination microscopy34 in the sdt-python library automatically matches the position of each marker in the donor emission channel with the position in the acceptor emission channel. A transformation T mapping the donor emission channel's coordinates to the acceptor emission channel's coordinates is found via a linear least-squares fit of an affine transformation to the markers' positions35. RANSAC is used to account for outliers, such as wrongly matched positions from the previous step. - Localize FRET probes independently upon donor and acceptor excitation in all frames and merge results into one table that contains the original frame number, 2-dimensional coordinates, and an identifier referring to the source image file.
NOTE: To this end, the software provides user interfaces to find appropriate options for the localization algorithm.- Localize FRET probes upon donor excitation in the sum of the images obtained from donor emission ImDD and acceptor emission ImDA, which hardly depend on the FRET efficiency. For information concerning the options for the localization algorithm, see section 4.
NOTE: Each sum image is calculated by transforming ImDD using the transformation T previously obtained from image registration and added pixelwise to ImDA. - Localize probes upon acceptor excitation in the acceptor emission channel ImAA (see section 4 for details about the localization algorithms).
- Localize FRET probes upon donor excitation in the sum of the images obtained from donor emission ImDD and acceptor emission ImDA, which hardly depend on the FRET efficiency. For information concerning the options for the localization algorithm, see section 4.
- Perform tracking and fluorescence intensity measurement.
- Specify the illumination sequence to permit the selection of donor and acceptor excitation frames, as well as frames for image segmentation from recorded image sequences.
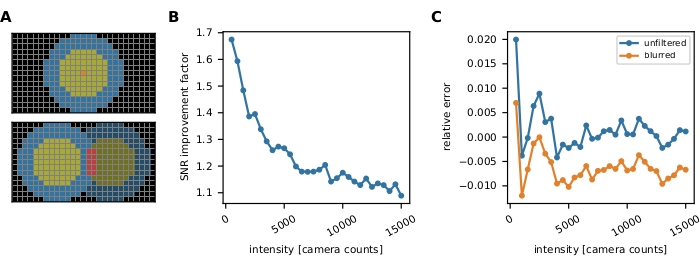
Figure 3: Single-molecule intensity measurement. (A) For a fluorophore located at the orange pixel, its uncorrected intensity Iuncorr is determined by summing up all pixels' intensities within a disk (yellow and orange pixels) large enough to cover all pixels affected by the signal: 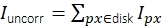 . The local background is computed as the mean of the pixels in a ring (blue pixels) around the disk:
. The local background is computed as the mean of the pixels in a ring (blue pixels) around the disk: 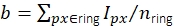 , where nring is the number of pixels in the ring. The fluorescence intensity I is the result of subtracting the background from the uncorrected intensity, I = Iuncorr – b × ndisk, where ndisk is the number of pixels in the disk. The circle radius is specified via the feat_radius parameter of the tracking method. The width of the ring is given by the bg_frame parameter. If the point spread function of one signal overlaps with the background ring of another (bottom panel), the affected pixels (red) are excluded from local background analysis. If two point spread functions overlap, fluorescence intensities cannot be calculated reliably and are therefore discarded. (B, C) Simulations show that applying a Gaussian blur with a standard deviation of 1 pixel improves the signal-to-noise ratio up to a factor of close to 2 at low fluorescence intensities (B) and introduces hardly any error (slight underestimation of less than 1%, (C))16. Moreover, the relative error (i.e., (Imeas – Itruth)/Itruth, where Itruth is the ground truth and Imeas is the outcome of the analysis) is constant over the whole intensity range and therefore cancels out for ratiometric quantities such as FRET efficiencies and stoichiometries. All plots are based on previously published work16. Abbreviations: SNR = signal-to-noise ratio; FRET = Förster resonance energy transfer. Please click here to view a larger version of this figure.
, where nring is the number of pixels in the ring. The fluorescence intensity I is the result of subtracting the background from the uncorrected intensity, I = Iuncorr – b × ndisk, where ndisk is the number of pixels in the disk. The circle radius is specified via the feat_radius parameter of the tracking method. The width of the ring is given by the bg_frame parameter. If the point spread function of one signal overlaps with the background ring of another (bottom panel), the affected pixels (red) are excluded from local background analysis. If two point spread functions overlap, fluorescence intensities cannot be calculated reliably and are therefore discarded. (B, C) Simulations show that applying a Gaussian blur with a standard deviation of 1 pixel improves the signal-to-noise ratio up to a factor of close to 2 at low fluorescence intensities (B) and introduces hardly any error (slight underestimation of less than 1%, (C))16. Moreover, the relative error (i.e., (Imeas – Itruth)/Itruth, where Itruth is the ground truth and Imeas is the outcome of the analysis) is constant over the whole intensity range and therefore cancels out for ratiometric quantities such as FRET efficiencies and stoichiometries. All plots are based on previously published work16. Abbreviations: SNR = signal-to-noise ratio; FRET = Förster resonance energy transfer. Please click here to view a larger version of this figure.
- Choose appropriate options for the trackpy36 algorithm used to link FRET probe localizations into trajectories. In particular, set the maximum search distance from one frame to the next and the number of consecutive frames for which a signal may go undetected, which can occur due to bleaching or missed localizations.
NOTE: These gaps are filled via interpolation between preceding and ensuing positions. These interpolated positions are marked and only used later for reading out intensity values but not for diffusion analysis. Tracks are analyzed in the coordinate system of the acceptor emission channel. For fluorescence intensity analysis (step 6.1.6.2), tracks are additionally transformed to the coordinate system of the donor emission channel using the inverse transformation T-1 obtained via image registration (see step 6.1.4). - Select options for the fluorescence intensity calculation algorithm (see Figure 3A for details). Specify i) the radius of a disk that, when centered on a signal's position, contains all pixels affected by that signal and ii) the width of a ring around each disk used to determine the local background.
NOTE: To reduce noise in the obtained intensity measurements, a Gaussian blur with a standard deviation of 1 pixel is applied to the images (Figure 3B,C).
- Use the analysis software functionality to process auxiliary image data from image sequences.
- Extract additional images recorded to facilitate segmentation (see step 2.2.1, marked by s in the excitation sequence (see Supplemental Information, section 1.2, step 3).
- Determine donor and acceptor excitation light profiles across the field of view from images recorded on a densely labeled sample (see step 3.2).
NOTE: The pixel-by-pixel mean is calculated from the images to compute the light profiles. The camera baseline is subtracted. Images are blurred using a Gaussian filter to reduce effects due to sample impurities. Finally, the resulting images are divided pixelwise by their maximum value to get the profile p(x,y) mapping coordinates onto the interval [0,1].
7. Visualization of FRET trajectories (optional)
- Use the inspector application to display single-molecule tracks in raw image data and the corresponding fluorescence intensities and apparent FRET efficiencies and stoichiometries.
NOTE: This is a valuable tool to assess the validity of chosen parameters and manually accept or reject individual time traces. See Supplemental Information for a screenshot and detailed usage information.
8. Analysis and filtering of single-molecule data (02. Analysis.ipynb)
- Use the 02. Analysis.ipynb Jupyter notebook for analysis and filtering of the single-molecule data obtained via the 01. Tracking.ipynb notebook. See the steps below for a typical analysis pipeline.
NOTE: Different scientific questions and experimental designs may require adjustments of the settings. The use of Jupyter notebooks permits easy adaption by omitting, rearranging, and amending analysis steps. For screenshots of each analysis step and description of function call parameters, refer to Supplemental Information.- Perform initial filtering steps.
- Discard signals with overlapping point spread functions as it is difficult to determine their fluorescence intensities reliably.
- In the case of inhomogeneous illumination, accept only signals located in well-illuminated regions within the field of view to ensure a good signal-to-noise ratio.
- If studying intramolecular FRET, restrict the analysis to those trajectories present from the beginning of the image sequence to ensure that all bleaching steps are recorded and can be properly evaluated later during the stepwise photobleaching analysis.
NOTE: When performing experiments with intermolecular FRET probes, wherein donor and acceptor fluorophores are not part of a preformed complex, it may not be feasible to restrict the analysis to initially present trajectories.
- Execute flatfield correction, which uses the excitation light source profiles obtained in step 6.1.7.2 to reverse the position-dependent fluorescence intensity variations caused by inhomogeneous illumination.
NOTE: The fluorescence intensity I(x,y) of a probe at the position (x,y) is corrected via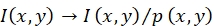 ; see Figure 1C,D.
; see Figure 1C,D. - Compute the apparent FRET efficiency Eapp (i.e., the fraction of energy transmitted from the donor fluorophore to the acceptor fluorophore) and the apparent stoichiometry Sapp (i.e., the number of donor fluorophores divided by the total number of fluorophores within a diffraction-limited spot).
NOTE: By plotting E vs. S for each data point, it is possible to distinguish alterations in the measured FRET efficiencies due to change in donor-acceptor distance from alterations due to changes in the stoichiometry18. This allows for the differentiation between E = 0 due to dye separation from E = 0 due to the absence of an active acceptor. E-S plots are used throughout the analysis as a tool for quality assessment; see Figure 4 as an example. - Perform stepwise analysis of photobleaching for the discrimination between single-molecular probes and aggregates. Choose to accept one of the following options.
NOTE: To this end, the analysis software applies a custom implementation32 of the changepoint detection algorithm PELT37 separately to the fluorescence intensity upon donor excitation (IDD + IDA) and acceptor excitation (IAA).- Choose option 1, wherein the acceptor fluorophore bleaches in a single step while the donor shows no partial bleaching (i.e., there is no bleaching step to non-zero intensity).
NOTE: This option further rejects trajectories where the donor bleaches before the acceptor in a single step. Option 1 is the preferred choice in case of high acceptor photobleaching rates. - Choose option 2, wherein the donor bleaches in a single step while there is no partial acceptor bleaching.
NOTE: This option further rejects trajectories where the donor bleaches after the acceptor in a single step. Option 2 is the preferred choice in case of high donor photobleaching rates. - Choose option 3, wherein either fluorophore bleaches in a single step while the other does not partially bleach.
NOTE: Option 3 gives higher flexibility than options 1 and 2 and would be the suggested preference for data analysis. - Choose option 4, wherein donor and acceptor fluorophores show single-step photobleaching or no photobleaching at all.
NOTE: Option 4 is preferred in case of low photobleaching rates.
- Choose option 1, wherein the acceptor fluorophore bleaches in a single step while the donor shows no partial bleaching (i.e., there is no bleaching step to non-zero intensity).
- Calculate the correction factors for donor emission leakage into the acceptor channel α, direct acceptor excitation δ, detection efficiencies γ, and excitation efficiencies β17.
- Use the correction factors to calculate the FRET efficiency E from the apparent efficiency Eapp and the stoichiometry S from the apparent stoichiometry Sapp.
- Perform further filtering steps. Select only data points from before the first bleaching event in each trajectory. Additionally, accept only trajectories with at least 75% of data points satisfying 0.35 < S < 0.6 to restrict the analysis to single-molecule probes (numbers are adjustable).
NOTE: The upper and lower bounds for should be chosen according to the spread of the population of interest versus the populations to be excluded from the analysis (e.g., donor-only and acceptor-only populations). Based on experience, 0.35 < S < 0.6 turned out to be a good choice for many experimental situations. - Perform image segmentation via global or adaptive thresholding methods35 on the appropriate auxiliary images (see steps 2.2.1 and 6.1.7) to restrict the analysis to distinct regions within the field of view.
NOTE: This permits, for instance, exclusive evaluation of probes located in a cell-SLB interface or on a patterned structure.
- Perform initial filtering steps.
9. Plotting of results and further analysis (03. Plot.ipynb)
NOTE: Refer to Supplemental Information for screenshots of the Jupyter notebook and description of function call parameters.
- Create E-S plots to verify that signals of incorrect stoichiometry have been correctly identified and removed.
- Plot histograms of FRET efficiencies to provide a well-established overview of the FRET efficiency distributions. Group the histograms for convenient comparison of results from different experiments.
- Evaluate the data further (e.g., diffusion analysis, conversion of FRET efficiencies to forces in experiments using molecular force sensors or transition analysis) within the notebook taking advantage of scientific Python libraries.
NOTE: Data can also be exported in many file formats as input to other analysis software.
A variety of low- and high-level information can be extracted from smFRET tracks depending on the scientific question of the experiment. Here, examples of analysis pipelines with analog and digital probes are presented: a peptide-based molecular force sensor16 and a DNA probe with stochastic switching of its conformation38, respectively. Refer to Figure 5 for the design and working principle of these probes.
After the localization and tracking algorithms have been executed as described in the protocol, the package offers multiple data visualization tools to optimize the initial parameters and subsequent filter steps: (i) visualization of individual smFRET events, (ii) optional image segmentation to analyze data in certain regions of interests, (iii) monitoring of filter steps via FRET efficiency vs. stoichiometry (E-S) plots. The visualization of the single-molecule data is presented in Figure 6.
Finally, the filtered FRET events are represented by an E-S plot and a FRET efficiency histogram (Figure 4). The E-S plot is a useful tool for optimizing the aforementioned filtering steps and investigating the final result. Partially bleached or incompletely labeled FRET sensors can be excluded by their stoichiometry value. Mobility parameters can be investigated by plotting an individual trajectory path in an x-y plot (Figure 6) or a mean square displacement (MSD) plot (Figure 4). The first method is especially useful for discriminating mobile from immobilized events, while the latter is used to calculate the diffusion coefficient.
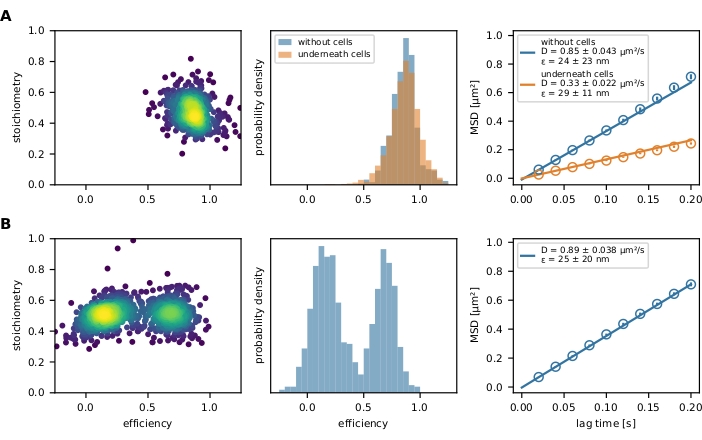
Figure 4: Exemplary output. (A) The FRET efficiency is plotted versus stoichiometry (E-S plot) for a population of the molecular force sensor (left panel) decorating a glass-supported lipid bilayer and strained by a T cell. Only one population cloud is visible. The respective histogram of FRET efficiencies exemplifies the difference between a force sensor population in presence and absence of cells (middle panel). No shift to lower FRET efficiencies of the sensor population in presence of T cells can be observed, indicating little to no force-dependent stretching of the sensor module. The MSD plot of these experimental conditions confirms that the force sensor population beneath a T cell moves considerably slower than their unbound counterparts (right panel). (B) The same analysis was performed with Holliday junction DNA sensor decorating a glass-supported fluid lipid bilayer. The E–S plot clearly shows two populations, which are also apparent in the FRET efficiency histogram. The MSD plot indicates the presence of one fast-moving sensor population. Abbreviations: FRET = Förster resonance energy transfer; MSD = mean square displacement. Please click here to view a larger version of this figure.
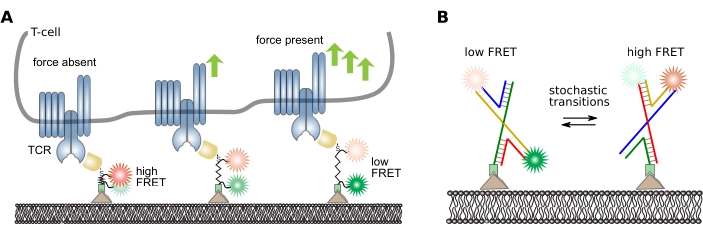
Figure 5: Design and working principle of intramolecular FRET probes. (A) Analog peptide sensor for quantification of mechanical molecular forces. The donor and acceptor fluorophores are covalently attached to either end of the peptide backbone. The sensor module is site-specifically attached to a specific ligand, which in turn binds a cell-resident surface receptor of interest (here, an antibody fragment specifically recognizing the beta chain of the T cell receptor). Upon receptor-ligand binding, force is exerted, and the sensor module extends and eventually recoils after bond cleavage. This panel is modified from 16. (B) Digital DNA sensor for quantification of FRET transitions. The FRET sensor is composed of four DNA strands forming a Holliday junction. The donor and acceptor fluorophore are covalently attached to two strands. Holliday junctions frequently switch their conformation depending on the surrounding buffer conditions. The stochastic switching of these conformations can be monitored by quantifying the FRET efficiency of individual probes. Abbreviations: TCR = T cell receptor; FRET = Förster resonance energy transfer. Please click here to view a larger version of this figure.
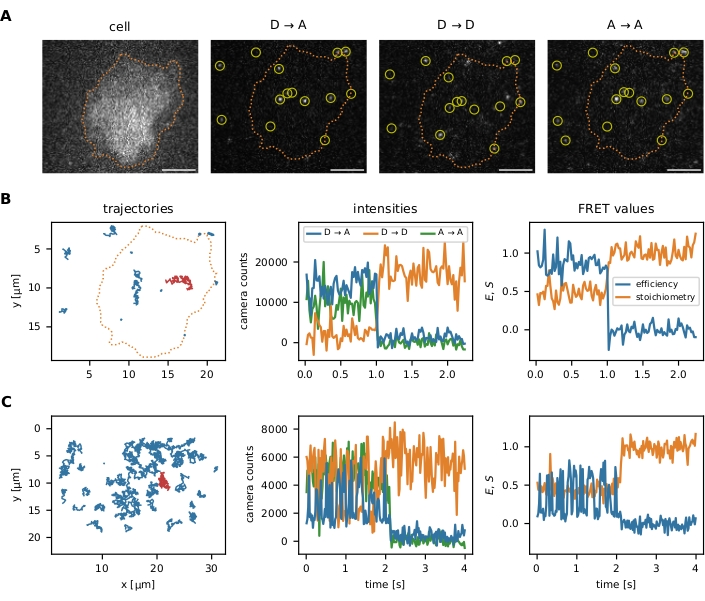
Figure 6: Examples of localization and tracking of FRET probes. (A) The FRET efficiency and stoichiometry of individual events are calculated by quantifying the intensity of the donor fluorophore upon donor excitation (D → D), the acceptor fluorophore upon donor excitation (D → A), and the acceptor fluorophore upon acceptor excitation (A → A). Nearest neighbor filtering prevents bias by overlapping point spread functions of close emitters. Image segmentation allows the user to choose certain smFRET events localized within an area of interest (e.g., a cell or a micropattern). As an example of image segmentation, T cells were stained with Fura-2 (displayed on the left) and subjected to adaptive thresholding to identify the cell edges (orange dotted line). Scale bars = 5 µm. (B) smFRET trajectories using the molecular force sensor. Individual trajectories can be plotted in the x-y plane, visualizing their diffusion behavior and localization (left panel). Furthermore, each trajectory's intensities can be plotted over time to identify FRET transitions or bleaching steps (middle panel shows the red trajectory from the left panel). The resulting FRET efficiency and stoichiometry can be visualized similarly (right panel). (C) smFRET trajectories using the Holliday junction DNA sensor. HBSS + 12 mM MgCl2 was used as a buffer during the measurements. Apart from the apparent acceptor bleaching step near the sequence end of these examples, the frequency of FRET transitions for each sensor can be determined. The Holliday junctions switch their conformation with a high frequency, whereas the molecular force sensor does not exhibit FRET transitions. This information makes it possible to adjust the experimental conditions, such as the delay between the frames, to increase or reduce the number of observed transitions. Abbreviations: FRET = Förster resonance energy transfer; smFRET = single-molecule FRET; HBSS = Hank's balanced salt solution. Please click here to view a larger version of this figure.
Supplemental Information: Localization and tracking of single molecules (01. Tracking.ipynb). Please click here to download this File.
