Single-Cell Factor Localization on Chromatin using Ultra-Low Input Cleavage Under Targets and Release using Nuclease
Summary
CUT&RUN and its variants can be used to determine protein occupancy on chromatin. This protocol describes how to determine protein localization on chromatin using single-cell uliCUT&RUN.
Abstract
Determining the binding locations of a protein on chromatin is essential for understanding its function and potential regulatory targets. Chromatin Immunoprecipitation (ChIP) has been the gold standard for determining protein localization for over 30 years and is defined by the use of an antibody to pull out the protein of interest from sonicated or enzymatically digested chromatin. More recently, antibody tethering techniques have become popular for assessing protein localization on chromatin due to their increased sensitivity. Cleavage Under Targets & Release Under Nuclease (CUT&RUN) is the genome-wide derivative of Chromatin Immunocleavage (ChIC) and utilizes recombinant Protein A tethered to micrococcal nuclease (pA-MNase) to identify the IgG constant region of the antibody targeting a protein of interest, therefore enabling site-specific cleavage of the DNA flanking the protein of interest. CUT&RUN can be used to profile histone modifications, transcription factors, and other chromatin-binding proteins such as nucleosome remodeling factors. Importantly, CUT&RUN can be used to assess the localization of either euchromatic- or heterochromatic-associated proteins and histone modifications. For these reasons, CUT&RUN is a powerful method for determining the binding profiles of a wide range of proteins. Recently, CUT&RUN has been optimized for transcription factor profiling in low populations of cells and single cells and the optimized protocol has been termed ultra-low input CUT&RUN (uliCUT&RUN). Here, a detailed protocol is presented for single-cell factor profiling using uliCUT&RUN in a manual 96-well format.
Introduction
Many nuclear proteins function by interacting with chromatin to promote or prevent DNA-templated activities. To determine the function of these chromatin-interacting proteins, it is important to identify the genomic locations at which these proteins are bound. Since its development in 1985, Chromatin Immunoprecipitation (ChIP) has been the gold standard for identifying where a protein binds to chromatin1,2. The traditional ChIP technique has the following basic workflow: cells are harvested and crosslinked (usually with formaldehyde), chromatin is sheared (usually with harsh sonication methods, necessitating crosslinking), the protein of interest is immunoprecipitated using an antibody that targets the protein (or tagged protein) followed by a secondary antibody (coupled to agarose or magnetic beads), crosslinking is reversed, protein and RNA are digested to purify DNA, and this ChIP enriched-DNA is used as the template for analysis (using radiolabeled probes1,2, qPCR3, microarrays5,6, or sequencing4). With the advent of microarrays and massively parallel deep sequencing, ChIP-chip5,6 and ChIP-seq4 have more recently been developed and allow for genome-wide identification of protein localization on chromatin. Crosslinking ChIP has been a powerful and reliable technique since its advent with major advances in resolution by ChIP-exo7 and ChIP-nexus8. In parallel to the development of ChIP-seq, native (non-crosslinking) protocols for ChIP (N-ChIP) have been established, which utilize nuclease digestion (often using micrococcal nuclease or MNase) to fragment the chromatin, as opposed to sonication performed in traditional crosslinking ChIP techniques9. However, one major drawback to both crosslinking ChIP and N-ChIP technologies has been the requirement for high cell numbers due to low DNA yield following the experimental manipulation. Therefore, in more recent years, many efforts have been toward optimizing ChIP technologies for low cell input. These efforts have resulted in the development of many powerful ChIP-based technologies that vary in their applicability and input requirements10,11,12,13,14,15,16,17,18. However, single-cell ChIP-seq based technologies have been lacking, especially for non-histone proteins.
In 2004, an alternative technology was developed to determine protein occupancy on chromatin termed Chromatin Endogenous Cleavage (ChEC) and Chromatin Immunocleavage (ChIC)19. These single-locus techniques utilize a fusion of MNase to either the protein of interest (ChEC) or to protein A (ChIC) for direct cutting of DNA adjacent to the protein of interest. In more recent years, both ChEC and ChIC have been optimized for genome-wide protein profiling on chromatin (ChEC-seq and CUT&RUN, respectively)20,21. While ChEC-seq is a powerful technique for determining factor localization, it requires developing MNase-fusion proteins for each target, whereas ChIC and its genome-wide variation, CUT&RUN, rely on an antibody directed toward the protein of interest (as with ChIP) and recombinant Protein A-MNase, where the Protein A can recognize the IgG constant region of the antibody. As an alternative, a fusion Protein A/Protein G-MNase (pA/G-MNase) has been developed that can recognize a broader range of antibody constant regions22. CUT&RUN has rapidly become a popular alternative to ChIP-seq for determining protein localization on chromatin genome-wide.
Ultra-low input CUT&RUN (uliCUT&RUN), a variation of CUT&RUN that enables the use of low and single-cell inputs, was described in 201923. Here, the methodology for a manual 96-well format single-cell application is described. It is important to note that since the development of uliCUT&RUN, two alternatives for histone profiling, CUT&Tag and iACT-seq have been developed, providing robust and highly parallel profiling of histone proteins24,25. Furthermore, scCUT&Tag has been optimized for profiling multiple factors in a single cell (multiCUT&Tag) and for application to non-histone proteins26. Together, CUT&RUN provides an attractive alternative to low input ChIP-seq where uliCUT&RUN can be performed in any molecular biology lab that has access to a cell sorter and standard equipment.
Protocol
Ethics statement: All studies were approved by the Institutional Biosafety Office of Research Protections at the University of Pittsburgh.
1. Prepare magnetic beads
NOTE: Perform prior to cell sorting and hold on ice until use.
- Pipette 30 µL of ConA-conjugated paramagnetic microspheres bead slurry mix per reaction to a fresh 1.5 mL microfuge tube and add 850 µL of Binding Buffer, pipetting gently to mix.
NOTE: ConA-conjugated paramagnetic microspheres are lectin-coated magnetic beads that permit lipid membrane binding. - Place the tube on a magnetic rack and allow the beads to magnetize for 1-2 min. Once the supernatant has cleared, remove and discard the supernatant without disturbing the beads.
- Remove the tube from the magnetic rack and wash the beads by resuspending in 1 mL of Binding Buffer.
- Repeat steps 1.2 and 1.3.
- Magnetize the beads for 2 min and remove the supernatant to discard.
- Remove the tube from the magnetic rack and resuspend the beads in 30 µL of Binding Buffer per reaction.
- Hold the washed bead mix on ice until cells are sorted.
2. Harvest cells
NOTE: This step is written for adhered cells and optimized for murine E14 embryonic stem cells. Culturing and harvesting the cells depend on the cell type.
- Remove the cells from the 37 °C incubator and examine them under a microscope to assure quality.
- Aspirate the media from the cell plate and rinse with 5 mL of 1x PBS.
- Aspirate PBS from the plate and harvest the cells (using traditional cell harvesting methods which will differ by cell type). Obtain single-cell suspension by gently pipetting up and down with a serological pipette against the culture dish, if necessary.
- Transfer the cell suspension to a 15 mL conical tube and spin down at 200 x g for 5 min.
- Aspirate off the media to discard and wash the cell pellet with 5 mL of PBS + 1% FBS.
- Spin down the cells at 200 x g for 5 min, discard the supernatant, and resuspend the cell pellet in 5 mL of PBS + 1% FBS.
- Count the cells and transfer 1 mL of 1 x 106 cells into a fresh 1.5 mL microfuge tube.
- Add 5 µL of 7-Amino-Actinomycin D (7-AAD), invert the tube well to mix, and then apply the sample to the cell sorter to sort live single cells into individual wells of a 96-well plate.
NOTE: 7-AAD dye is excluded from live cells, and therefore can be used in live-cell sorting.
3. Cell sorting and lysis
- Prepare a cell sorter-compatible 96-well plate with 100 µL of Nuclear Extraction (NE) Buffer in each well prior to cell sorting.
- Sort the cells into 96-well plates following the manufacturer's instructions.
- Quickly spin the plate (600 x g for 30 s) to assure cells are in the buffer within the wells.
NOTE: It is worth testing in a preliminary experiment whether the cells being used are reliably brought to the bottom of the wells. - Hold the samples on ice for 15 min.
- Spin down the samples at 600 x g for 5 min at 4 °C and carefully pipette to remove the supernatant (leaving behind 5 µL).
- Resuspend each sample in 55 µL of NE buffer and add 30 µL of the prewashed ConA-conjugated paramagnetic microspheres (from step 1.7; in Binding Buffer) to each reaction.
- Incubate at room temperature for 10 min.
4. Pre-block samples to prevent early digestion by MNase
- Place the plate on a 96-well magnetic rack, allow the beads to bind for a minimum of 5 min, and then remove and discard the supernatant.
- Add 100 µL of Blocking Buffer to the nucleus-bound beads and mix with gentle pipetting.
- Incubate for 5 min at room temperature.
5. Addition of primary antibody
- Place the plate on a 96-well magnetic rack, allow the supernatant to clear for a minimum of 5 min, and then remove and discard the supernatant without disturbing the beads.
- Remove the plate from the magnetic rack and resuspend the beads in 100 µL of Wash Buffer per reaction with gentle pipetting.
- Place the plate back on the 96-well magnetic rack, allow the supernatant to clear, and then remove and discard the supernatant.
- Resuspend the beads in 25 µL of Wash Buffer per reaction with gentle pipetting.
- Make a primary antibody master mix: 25 µL of Wash Buffer + 0.5 µL of antibody per reaction.
- While gently vortexing the nuclei-bound beads, add 25 µL of the primary antibody master mix to each sample being treated with an antibody targeting the protein of interest (typically 1:100 final dilution). Add 25 µL of Wash Buffer with no antibody, if performing a control.
- Incubate for 1 h at room temperature.
- Place the samples on a 96-well magnetic rack, allow the supernatant to clear for a minimum of 5 min, and then remove and discard the supernatant without disturbing the beads.
- Remove the plate from the magnetic rack and wash the beads with 100 µL of Wash Buffer, resuspending by pipetting.
6. Addition of pA-MNase or pA/G-MNase
NOTE: Protein A has a high affinity for IgG molecules from certain species such as rabbits but is not suitable for IgGs from other species such as mice or rats. Alternatively, Protein A/G-MNase can be used. This hybrid binds rabbit, mouse, and rat IgGs, avoiding the need for secondary antibodies when mouse or rat primary antibodies are used.
- Place the plate back on a 96-well magnetic rack, allow the supernatant to clear for a minimum of 5 min, and then remove and discard the supernatant without disturbing the beads.
- Remove the plate from the magnetic rack and resuspend each sample in 25 µL of Wash Buffer.
- Make a pA-MNase master mix (25 µL of Wash Buffer + optimized amount of pA-MNase per reaction).
- While gently vortexing, add 25 µL of the pA-MNase master mix to each sample, including the control samples.
NOTE: The concentration of pA-MNase varies upon preparation, if homemade, and should be tested prior to use upon each independent purification. For pA/G-MNase, 2.5 µL of the 20x stock should be used. - Incubate the samples for 30 min at room temperature.
- Place the plate on a 96-well magnetic rack, allow the supernatant to clear for a minimum of 5 min, and then remove and discard the supernatant without disturbing the beads.
- Remove the plate from the magnetic rack and wash the beads with 100 µL of Wash Buffer, resuspending by gentle pipetting.
7. Directed DNA digestion
- Place the plate on a 96-well magnetic rack, allow the supernatant to clear for a minimum of 5 min, and then remove and discard the supernatant.
- Remove the samples from the magnetic rack and resuspend the beads in 50 µL of Wash Buffer by gentle pipetting.
- Equilibrate the samples to 0 °C in an ice/water mixture for 5 min.
- Remove the samples from the 0 °C ice/water bath and add 1 µL of 100 mM CaCl2 using a multichannel pipette. Mix well (3-5 times) with gentle pipetting using a larger volume multichannel pipette, and then return the samples to 0 °C.
NOTE: Mixing well here is essential. CaCl2 is added to activate MNase digestion of DNA flanking the protein of interest. - Start a 10 min timer as soon as the plate is back in the ice/water bath.
- Stop the reaction by pipetting 50 µL of 2XRSTOP+ Buffer into each well, in the same order as the CaCl2 was added.
NOTE: Make 2XRSTOP+ Buffer before the 10 min digestion is over to prevent over digestion.
8. Sample fractionation
- Incubate the samples for 20 min at 37 °C.
- Spin the plate at 16,000 x g for 5 min at 4 °C.
- Place the plate on a 96-well magnetic rack, allow the supernatant to clear for a minimum of 5 min, and transfer supernatants to a fresh 96-well plate. Discard the beads.
9. DNA extraction
- Add 1 µL of 10% Sodium Dodecyl Sulfate (SDS) and 0.83 µL of 20 mg/mL Proteinase K to each sample.
CAUTION: SDS powder is harmful if inhaled. Users should use in well-ventilated spaces wearing goggles, gloves, and an N95-grade respirator, handling with care. - Mix the samples by gentle pipetting.
- Incubate the samples for 10 min at 70 °C.
- Return the plate to room temperature and add 46.6 µL of 5 M NaCl and 90 µL of 50% PEG 4000. Mix by gentle pipetting.
- Add 33 µL of polystyrene-magnetite beads to each sample and incubate for 10 min at room temperature.
NOTE: Be sure to bring polystyrene-magnetite beads to room temperature (~30 min) and mix well before using. - Place the plate on a magnetic rack and allow the supernatant to clear for ~5 min, and then carefully discard the supernatant without disturbing the beads.
- Rinse 2x with 150 µL of 80% ethanol without disturbing the beads.
CAUTION: Ethanol is highly flammable and causes skin, eye, and lung irritation. Perform this step with appropriate laboratory clothing and in a vented hood. - Spin the plate briefly at 1000 x g for 30 s. Place the plate back on a 96-well magnetic rack and remove all residual EtOH without disturbing the beads.
- Air-dry the samples for ~2-5 min.
NOTE: Do not dry the beads for longer than 5 min. If diligent about removing the EtOH, 2-3 min of drying is sufficient. - Resuspend the beads with 37.5 µL of 10 mM Tris-HCl (pH 8) and incubate for 5 min at room temperature.
- Place the plate on a magnetic rack and allow the beads to bind for 5 min.
- Transfer 36.5 µL of the supernatant to a fresh thermocycler compatible 96-well plate. Discard the beads.
NOTE: The experiment can be stopped here by storing samples at -20 °C or can continue with the library build (steps 10-15).
10. End repair, phosphorylation, adenylation
NOTE: The reagents are sourced as referenced in the Table of Materials. The below protocol follows a similar method to the commercial kit such as NEBNext Ultra DNA II kit.
- Dilute 5 U/µL of T4 DNA polymerase 1:20 in 1x T4 DNA ligase buffer.
- Prepare an end-repair/3'A master mix: 2 µL of 10x T4 DNA ligase buffer, 2.5 µL of 10 mM dNTPs, 1.25 µL of 10 mM ATP, 3.13 µL of 40% PEG 4000, 0.63 µL of 10 U/µL T4 PNK, 0.5 µL of diluted T4 DNA polymerase, 0.5 µL of 5U/µL Taq DNA polymerase, with a total volume of 13.5 µL per reaction.
NOTE: Be sure to bring 40% PEG 4000 to room temperature before pipetting. - Add 13.5 µL of end-repair/3'A master mix to 36.5 µL of DNA.
- Mix the reaction by quick vortex, and then a quick spin (500 x g for 10 s).
- Incubate using the following reaction conditions in a pre-cooled thermocycler with a heated lid for temperatures >20 °C. Use the reaction conditions: 12 °C for 15 min, 37 °C for 15 min, 72 °C for 20 min, hold at 4 °C.
11. Adapter ligation
NOTE: Keep the samples on ice while setting up the following reaction. Allow ligase buffer to come to room temperature before pipetting. Dilute the Adaptor (see Table of Materials) in a solution of 10 mM Tris-HCl containing 10 mM NaCl (pH 7.5). Due to the low yield, do not pre-quantify the CUT&RUN-enriched DNA. Rather, generate 25-fold dilutions of the adaptor, using a final working adaptor concentration of 0.6 µM.
- Make a ligation master mix: 55 µL of ligase buffer (2x), 5 µL of T4 DNA ligase, and 5 µL of diluted Adaptor, with a total volume of 65 µL per reaction.
- Add 65 µL of the master ligation mix to 50 µL of DNA from step 10.5.
- Mix by quick vortexing, followed by quick spinning (500 x g for 10 s).
- Incubate at 20 °C in a thermocycler (without a heated lid) for 15 min.
NOTE: Proceed immediately to the following step.
12. USER digestion
- Add 3 µL of USER enzyme to each sample, vortex, and spin (500 x g for 10 s).
- Incubate in thermal cycler at 37 °C for 15 min (heated lid set to 50 °C).
13. Polystyrene-magnetite bead clean-up following ligation reaction
NOTE: Allow polystyrene-magnetite beads to equilibrate at room temperature (~30 min). Vortex to homogenize the bead solution before using. Perform the following steps at room temperature.
- Add 39 µL (0.33x) of polystyrene-magnetite bead solution to each well containing adaptor-ligated DNA.
- Mix thoroughly by pipetting, and then incubate the samples at room temperature for 15 min to allow DNA to bind to the beads.
- Place the samples on a 96-well magnetic rack and incubate for 5 min until the supernatant is clear.
- Keep the plate on the magnetic rack and carefully remove and discard the supernatant without disturbing the beads.
- Rinse the beads with 200 µL of 80% EtOH without disturbing the beads.
- Incubate for 30 s on a 96-well magnetic rack to allow the solution to clear.
- Remove and discard the supernatant without disturbing the beads.
- Repeat steps 13.5-13.7 for a total of two washes.
- Spin the plate briefly at 500 x g for 10 s, place the plate back on a 96-well magnetic rack, and remove residual EtOH without disturbing the beads.
- Keep the plate on the magnetic rack and air-dry the samples for 2 min.
NOTE: Do not over-dry the beads. - Remove the plate from the magnetic rack and resuspend the beads in 28.5 µL of 10 mM Tris-HCl (pH 8).
CAUTION: Hydrochloric acid is very corrosive. Users should handle it with care in a chemical fume hood wearing goggles, gloves, and a lab coat. - Thoroughly resuspend beads by pipetting, taking care to not produce bubbles.
- Incubate for 5 min at room temperature.
- Place the plate on a 96-well magnetic rack and allow the solution to clear for 5 min.
- Transfer 27.5 µL of supernatant to a new PCR plate and discard the beads.
14. Library enrichment
NOTE: Primers are diluted with the same solution as the adaptor. For this library build, use a final working primer concentration of 0.6 µM.
- Add 5 µL of diluted indexed primer (see Table of Materials) to each sample.
NOTE: Each sample needs a different index to be identified after sequencing. - Prepare a PCR master mix: 10 µL of 5x high fidelity PCR buffer, 1.5 µL of 10 mM dNTPs, 5 µL of diluted Universal primer, 1 µL of 1 U hot start high fidelity polymerase, with a total master mix volume of 17.5 µL per sample.
- Add 17.5 µL pf PCR mix to 32.5 µL of purified adaptor-ligated DNA (5 µL of indexed primer is included in this volume).
- Mix the solution by pipetting.
- Incubate in a thermocycler using the following reaction conditions with a maximum ramp rate of 3 °C/s: 98 °C for 45 s, 98 °C for 45 s, 60 °C for 10 s, repeat the second and third steps 21 times, 72 °C for 1 min, hold at 4 °C.
NOTE: The samples can be kept at 4 °C for short-term storage or -20 °C for long-term storage.
15. Polystyrene-magnetite bead clean-up
- Add 60 µL (1.2x) of polystyrene-magnetite beads to each sample.
- Resuspend the beads by pipetting and incubate for 15 min at room temperature.
- Place the plate on a magnetic rack for 5 min until the solution is clear.
- Discard the supernatant and rinse the beads with 200 µL of 80% EtOH without disturbing the beads.
- Repeat the wash step for a total of two 80% EtOH washes.
- Spin the plate at 500 x g for 10 s, place the plate on a 96-well magnetic rack, and allow the beads to bind for 5 min.
- Pipette to remove excess EtOH without disturbing the beads and allow the beads to air-dry for 2 min.
NOTE: Do not over-dry the beads. - Resuspend the beads in 21 µL of nuclease-free water and incubate for 5 min at room temperature.
- Place the plate on a magnetic rack and allow the solution to clear for 5 min.
- Transfer 20 µL of the supernatant to a new plate.
NOTE: The experiment can be stopped here by storing the sample at -20 °C. - Quantify library concentrations with a fluorometer (see Table of Materials), using a 1x HS reagent.
- If the concentration allows, run 30 ng of sample on 1.5% agarose gel with a low molecular weight ladder to visualize. Alternatively, visualize on a Fragment Analyzer or related instrument.
- Sequence libraries on an Illumina platform to obtain ~50,000-100,000 uniquely mapped reads.
Representative Results
Here, a detailed protocol is presented for single-cell protein profiling on chromatin using a 96-well manual format uliCUT&RUN. While results will vary based on the protein being profiled (due to protein abundance and antibody quality), cell type, and other contributing factors, anticipated results for this technique are discussed here. Cell quality (cell appearance and percent of viable cells) and single-cell sorting should be assessed prior to or at the time of live-cell sorting into the NE buffer. An example of ES cell colonies and cell sorting is shown in Figure 2A,B. Specifically, low-quality cells should not be used, and if the quality is an issue, care should be taken to follow guidelines for the specific cell type. In addition, accurate cell sorting by the cell sorting instrument should be assessed in advance of experimentation. For example, test cells could be sorted and stained using Hoechst 33342 stain and counted to assure either 0 or 1 cell is found in each well. If single cells are not found, sorting conditions must be optimized. After library preparation, samples can be assessed on either an agarose gel (if the concentration permits for loading of 30 ng or more, as there is a lower limit to DNA visualization on an agarose gel) or a Fragment Analyzer (or similar device such as a Tapestation or Bioanalyzer) prior to sequencing and example results are shown in Figure 2C,D. Specifically, the expected size distribution is from ~150 to ~500 bp. In higher cell amounts, CUT&RUN performed on large proteins (such as histones) will have a right-sided size distribution, where the majority of DNA will be seen ~270 bp; however, this shoulder is typically not observed in single-cell experiments.
After sequencing, the quality of the sequencing reads should be assessed using FASTQC. The percent of uniquely mapping reads should be determined. Typically, 0.5%-10% uniquely mapping reads are observed in single-cell experiments. These mapping percentages are similar to other DNA-based single-cell techniques30. Next, the size distribution of the reads after mapping should be determined to assure the profile is similar to pre-sequencing (with the adaptor sequence no longer contributing to the read sizes).
After data quality has been assessed, protein occupancy can be visualized using various methods: single locus genome browser images can be visualized using UCSC genome browser or IGV (Figure 3A) and genome-wide occupancy patterns over specific genomic coordinates can be visualized using metaplots (Figure 3B, bottom), heatmaps (Figure 3B, top), or 1D heatmaps (Figure 3C). For more information on data analysis, refer to the study by Patty and Hainer27. Single-cell data from a diploid cell will result in up to four reads contributing to each locus (four reads if the cell was in mitosis), but more often one or two reads. Therefore, the data is binary, and a high background can be more easily mistaken for occupancy, relative to high cell experiments. Therefore, it is recommended to perform a parallel CUT&RUN experiment on a high cell number (5,000 to 100,000 cells), if possible, to acquire all the possible binding locations for the protein of interest. Then, single-cell data can be compared to possible binding locations. In the examples shown in Figure 3, single-cell CTCF uliCUT&RUN results are compared to high cell CTCF uliCUT&RUN (Figure 3A) or ChIP-seq (Figure 3B,C). As demonstrated previously, CTCF, SOX2, and NANOG single-cell uliCUT&RUN peaks largely represented stronger peaks from high cell ChIP-seq datasets23.
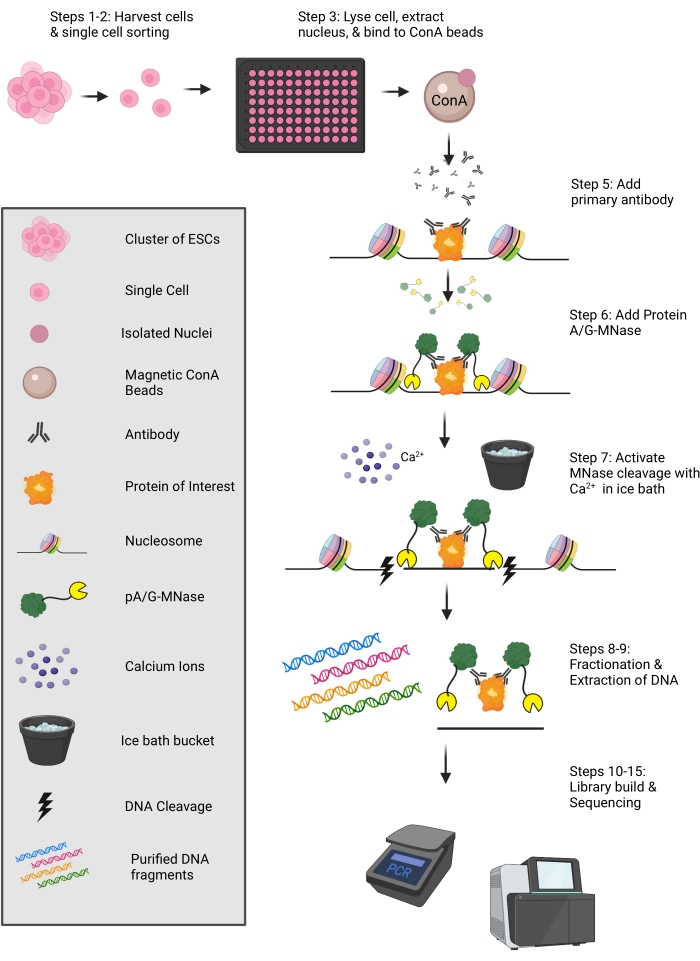
Figure 1: Schematic of the uliCUT&RUN protocol. Cells are harvested and sorted into a 96-well plate containing NE buffer. Individual nuclei are then bound to ConA-conjugated paramagnetic microspheres, and sequentially an antibody (targeting the protein of interest) and pA-MNase or pA/G-MNase are added. Protein-adjacent DNA is cleaved via MNase, and DNA is then purified for use in library preparation. This figure was created with Biorender.com. Please click here to view a larger version of this figure.
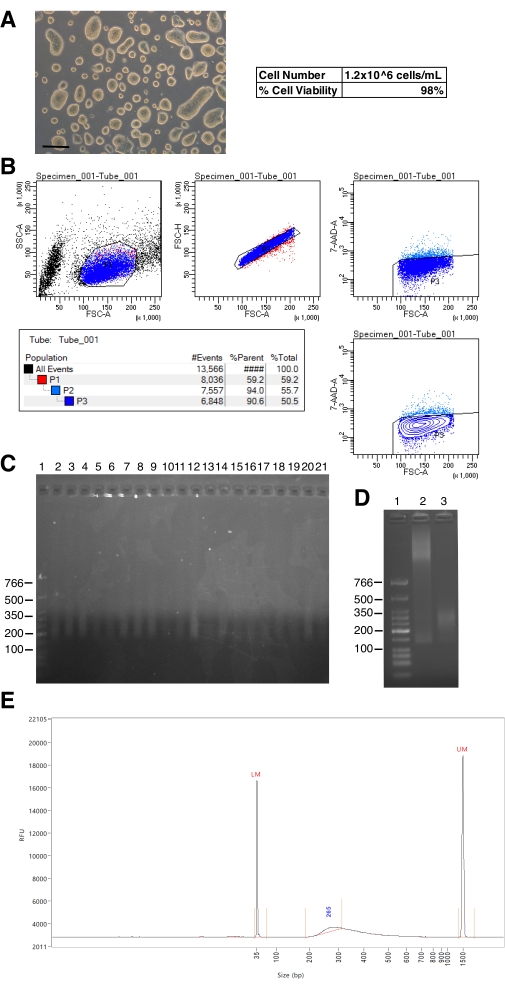
Figure 2: Example results from cell sorting and quality control of uliCUT&RUN libraries. (A) Image of high-quality murine embryonic stem (ES) cells. Scale bar: 200 µm. (B) Output from single-cell sorting after adding 7AAD to harvested ES cells and sorting on a FACS instrument. (C) Ethidium bromide-stained agarose gel depicting completed uliCUT&RUN libraries. Lane 1 is a low molecular weight ladder and lanes 2-21 are examples of successful individual single-cell uliCUT&RUN libraries prior to sequencing. (D) Ethidium bromide-stained agarose gel depicting sub-optimal and optimal completed uliCUT&RUN libraries. Lane 1 is a low molecular weight ladder, lane 2 is a sub-optimal library due to inefficient MNase digestion, and lane 3 is a successful library with appropriate digestion. (E) Fragment analyzer distribution of one single cell uliCUT&RUN library prior to sequencing. Please click here to view a larger version of this figure.
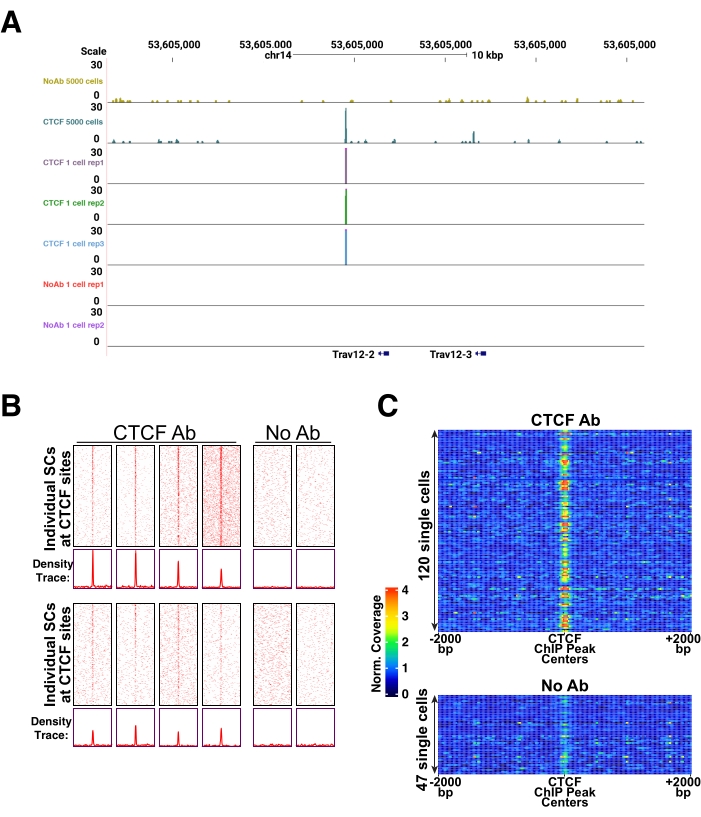
Figure 3: Example of expected results for single ES cell uliCUT&RUN data. (A) Browser track of high cell number (5,000 cells) CTCF or negative control (No Antibody, No Ab) uliCUT&RUN (top two tracks) and single-cell CTCF uliCUT&RUN. The image is reproduced, with permission, from Patty and Hainer27. (B) Heatmaps (top) and metaplots (bottom) of single-cell CTCF or negative control (No Ab) uliCUT&RUN over previously published CTCF ChIP-seq sites (GSE11724). The image is reproduced, with permission, from Hainer et al.23. (C) 1D heatmaps of single-cell CTCF or negative control (No Ab) uliCUT&RUN over previously published CTCF ChIP-seq sites (GSE11724). The image is reproduced, with permission, from Hainer et al.23. Please click here to view a larger version of this figure.
Table 1: Composition of various buffers used in this protocol. Volume of stock solution required is listed with final concentration written in parentheses. Please click here to download this Table.
Discussion
CUT&RUN is an effective protocol to determine protein localization on chromatin. It has many advantages relative to other protocols, including: 1) high signal-to-noise ratio, 2) rapid protocol, and 3) low sequencing read coverage required thus leading to cost savings. The use of Protein A- or Protein A/G-MNase enables CUT&RUN to be applied with any available antibody; therefore, it has the potential to quickly and easily profile many proteins. However, adaptation to single-cell for any protein profiling on chromatin has been difficult, especially when compared to single-cell transcriptomics (i.e., scRNA-seq), due to the low copy number of DNA relative to RNA (two to four copies of DNA versus possible thousands of RNA copies).
In the protocol detailed above, several critical steps should be considered. First, the appropriate sorting of cells is dependent on the cell (or tissue) type, and care should be taken for accurate sorting. It is recommended to test how effective single-cell sorting is with the instrument in advance of any experiment. Second, effective antibody choice is essential. Before proceeding to a single-cell application, it is recommended to test the antibody in a high cell number experiment (as well as other standard antibody tests, such as confirming specificity using western blot after knockdown, titration of the antibody in CUT&RUN experiments, etc.). Third, use a negative control, such as IgG or no primary antibody, because an appropriate comparison to the experimental samples is essential for the interpretation of the data quality and biological results. When comparing single-cell experimental results to a high cell number dataset, the negative control single-cell experiments should have less read coverage over those binding sites identified in the high cell experiment and rather have a random distribution of reads across the genome (with a bias for open regions of chromatin). Fourth, care should be taken when adding and activating the Protein A- or Protein A/G-MNase so as not to over digest the chromatin: do not overheat the samples with your hands, maintain the samples in an ice/water-bath temperature (0 °C), and chelate the reaction at the appropriate time. Fifth, care should be taken throughout the uliCUT&RUN experiment and library preparation, due to low material. For example, extended incubation on the magnetic rack during bead binding to assure the supernatant has cleared and taking care not to disturb the beads when removing the supernatant are essential for sufficient yield. Sixth, depending on the questions being addressed, the number of single-cell experiments being performed is an important consideration. Some of the single-cell experiments will fail (as with all low-input experiments), and, therefore, the number of positive experimental results required for appropriate interpretation should be considered in advance of beginning the experiment. The number of cells to include in the experiment is dependent on the amount of experimental data required by the investigator and the quality of the antibody. Finally, note that equivalent results could be achieved with reduced volumes of many aspects of this protocol. Steps where the volume was reduced by 50% include the volume of NE buffer, wash buffer, primary antibody mixture (including the amount of primary antibody), pA-MNase mixture (including the amount of pA-MNase), and all steps in the library preparation.
Based on the complicated nature of factor profiling on chromatin, there are many potential sources of issues and places where troubleshooting may become necessary. While there may be many steps where issues can arise, three major issues have been observed: 1) low DNA yield for input into library build; 2) high background signal in experimental samples, and 3) low yield after library build. If there is insufficient DNA for library preparation and sequencing (point 1), note the following troubleshooting advice: a) there may have been incomplete membrane lysis and therefore the lysis time with NE buffer can be increased; b) there may have been inefficient binding of the nucleus to the ConA-conjugated paramagnetic microspheres and this could be remedied through appropriate mixing upon addition of these beads; c) there may have been too little antibody added, and therefore it is recommended to perform a titration of antibody to identify the most effective amount; d) incubation times with either the primary antibody or the Protein A- or Protein A/G-MNase are either too short (i.e., not enough time to permit binding) or too long (these are native, uncrosslinked samples), and could be optimized; e) the interaction of the target protein with chromatin could be too transient to capture in native conditions and therefore crosslinking CUT&RUN could be performed28. While single-cell datasets will yield high background, there may be excessively high background where the signal is hard to interpret from the background (point 2). For this issue, note the following advice: a) the blocking step with EDTA to prevent pre-emptive MNase digestion could be increased or optimized; b) if there is excessive cutting, it could be due to having too much Protein A- or Protein A/G-MNase, and therefore a titration of appropriate amounts can be performed; c) over digestion by MNase could result in the high background, and therefore the appropriate mixing of calcium chloride upon addition and optimization for MNase digestion time should be assessed. Finally, efficient uliCUT&RUN-enriched DNA may have been recovered, but a low amount of library may be recovered (point 3). For this issue, the following are recommended: a) appropriate handling and use of polystyrene-magnetite beads to assure the correct purification and no DNA loss; specifically, it is recommended to have 15 min incubations and a minimum of 5 min to magnetize beads to prevent loss; b) under-amplification of the library at the PCR stage would result in low yield and therefore the appropriate cycles should be determined using qPCR (as previously established for ATAC-seq libraries29).
As with all technologies, there are limitations to uliCUT&RUN that should be considered before initiating any experimentation. First, these experiments are designed and optimized for native conditions, and therefore if a protein is only transiently interacting with chromatin, a crosslinking approach may be necessary to ensure recovery of the interaction. Second, as with all antibody-based techniques, the quality of the antibody is important. Care should be taken to assure the quality and consistency of the antibody in advance of undertaking any experiments. Third, MNase background cutting can occur and, while there is a consistently lower background signal in CUT&RUN relative to other experiments, the background signal can be high in single-cell experiments and therefore appropriate controls and analyses should be performed. While the binary nature of single-cell profiling data limits the visualization, more advanced computational genomic technologies, such as dimensional reduction and others can be performed (as previously described27). Finally, while single-cell profiling has been expanded here to 96-well format, this is low throughput relative to other single-cell technologies that utilize 10xGenomics or other formats.
Tethering-based profiling technologies such as ChEC-seq20, CUT&Tag24, CUT&RUN21, and uliCUT&RUN23, can determine factor localization on chromatin with a faster experimental timeline, lower background, and lower cost than traditional profiling technologies, such as ChIP-seq. Therefore, these are very exciting technologies for application to precious samples such as patient samples or early developmental samples. Furthermore, application to single cells can provide complementary studies performed using other single-cell experiments such as scRNA-seq and scATAC-seq30. As described using these more broadly used single-cell technologies, novel insights can be gained relative to bulk cell experiments. Single-cell protein profiling on chromatin is anticipated to become more regularly used as the technologies continue to improve and permit more parallelization.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
We thank members of the Hainer Lab for reading and comments on an earlier version of this manuscript. This project used the NextSeq500 available at the University of Pittsburgh Health Sciences Sequencing Core at UPMC Children's Hospital of Pittsburgh for sequencing with special thanks to its director, William MacDonald. This research was supported in part by the University of Pittsburgh Center for Research Computing through the computer resources provided. This work was supported by the National Institutes of Health Grant Number R35GM133732 (to S.J.H.).
Materials
| 1.5 mL clear microfuge tubes | ThermoFisher Scientific | 90410 | |
| 1.5 mL tube magnetic rack | ThermoFisher Scientific | 12321D | |
| 1.5 mL tube-compatible cold centrifuge | Eppendorf | 5404000537 | |
| 10 cm sterile tissue culture plates | ThermoFisher Scientific | 150464 | |
| 10X T4 DNA Ligase buffer | New England Biolabs | B0202S | |
| 15 mL conical tubes | VWR | 89039-656 | |
| 1X TE buffer | ThermoFisher Scientific | 12090015 | |
| 200 µL PCR tubes | Eppendorf | 951010022 | |
| 2X quick ligase buffer | New England Biolabs | M2200 | Ligase Buffer |
| 5X KAPA HiFi buffer | Roche | 7958889001 | 5X high fidelity PCR buffer |
| 7-Amino-Actinomycin D (7-AAD) | Fisher Scientific | BDB559925 | |
| 96-well magnetic rack | ThermoFisher Scientific | 12027 or 12331D | |
| 96-well plate | VWR | 82006-636 | |
| AMPure XP beads | Beckman Coulter | A63881 | polystyrene-magnetite beads; Due to potential variability between AMPure XP bead lots, it is recommended that your AMPure bead solution be calibrated. See manufacturer’s instructions |
| Antibody to protein of interest | varies | ||
| ATP | ThermoFisher Scientific | R0441 | |
| BioMag Plus Concanavalin A beads | Polysciences | 86057-10 | ConA-conjugated paramagnetic microspheres |
| BSA | |||
| Calcium Chloride (CaCl2) | Fisher Scientific | AAJ62905AP | |
| Cell sorter | BD FACSAria II cell sorter | Requires training | |
| Cell-specific media for cell culture | Varies | ||
| Chloroform | ThermoFisher Scientific | C298-500 | Chloroform is a skin irritant and harmful if swallowed; handle in a chemical fume hood using gloves, a lab coat, and goggles |
| Computer with 64-bit processer and access to a super computing cluster | For computational analyses of resulting sequencing datasets | ||
| DNA spin columns | Epoch Life Sciences | 1920-250 | |
| dNTP set | New England Biolabs | N0446S | |
| EGTA | Sigma Aldrich | E3889 | |
| Electrophoresis equipment | varies | ||
| Ethanol | Fisher Scientific | 22032601 | 100% vol/vol ethanol is highly flammable; handle in a chemical fume hood using gloves, a lab coat, and goggles |
| Ethylenediaminetetraacetic acid (EDTA) | Fisher Scientific | BP2482100 | |
| FBS | Sigma Aldrich | F2442 | |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycogen | VWR | 97063-256 | |
| HEPES | Fisher Scientific | BP310-500 | |
| Heterologous S. cerevisiae DNA spike-in | homemade | Prepared from crosslinked, MNase-digested, and agarose gel extracted genomic DNA purified of protein/RNA and diluted to 10 ng/mL. We recommend yeast genomic DNA, but other organisms can be used if needed. | |
| Hydrochloric Acid (HCl) | Fisher Scientific | A144-212 | Hydrochloric Acid is very corrosive; handle in a chemical fume hood using gloves, a lab coat, and goggles |
| Ice Bucket | varies | ||
| Illumina Sequencing platform (e.g., NextSeq500) | Illumina | ||
| Incubator with temperature and atmosphere control | ThermoFisher Scientific | 51030284 | |
| KAPA HotStart HiFi DNA Polymerase with 5X KAPA HiFi buffer | Roche | 7958889001 | hotstart high fidelity polymerase |
| Laminar flow hood | Bakery Company | SG404 | |
| Manganese Chloride (MnCl2) | Sigma Aldrich | 244589 | |
| Micropipette set | Rainin | 30386597 | |
| Minifuge | Benchmark Scientific | C1012 | |
| NEB Adaptor | New England Biolabs | E6612AVIAL | Adaptor |
| NEB Universal primer | New England Biolabs | E6611AVIAL | Universal Primer |
| NEBNext Multiplex Oligos for Illumina kit | New England Biolabs | E7335S/L, E7500S/L, E7710S/L, E7730S/L | Indexed Primers |
| Negative control antibody | Antibodies-Online | ABIN101961 | |
| Nuclease Free water | New England Biolabs | B1500S | |
| PCR thermocycler | Eppendorf | 2231000666 | |
| Phase lock tubes | Qiagen | 129046 | |
| Phenol/Chloroform/Isoamyl Alcohol (PCI) | ThermoFisher Scientific | 15593049 | Phenol is harmful if swallowed or upon skin contact; handle in a chemical fume hood using gloves, a lab coat, and goggles |
| Phsophate buffered saline (PBS) | ThermoFisher Scientific | 10814010 | |
| Pipette aid | Drummond Scientific | # 4-000-100 | |
| Polyethylene glycol (PEG) 4000 | VWR | A16151 | |
| Potassium Chloride (KCl) | Sigma Aldrich | P3911 | |
| Potassium Hydroxide (KOH) | Fisher Scientific | P250-1 | CAUTION KOH is an eye/skin irritant as a solid and corrosive in solution. Handle in a chemical fume hood using gloves, a lab coat, and goggles |
| Protease Inhibitors | ThermoFisher Scientific | 78430 | |
| ProteinA/G-MNase | Epicypher | 15-1016 | pA/G-MNase |
| ProteinA-MNase, purified from pK19pA-MN | Addgene | 86973 | |
| Proteinase K | New England Biolabs | P8107S | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay tubes | ThermoFisher Scientific | Q32856 | |
| Qubit Fluorometer | ThermoFisher Scientific | Q33238 | |
| Quick Ligase with 2X Quick Ligase buffer | New England Biolabs | M2200S | |
| RNase A | New England Biolabs | T3010 | |
| Sodium Acetate (NaOAc) | ThermoFisher Scientific | BP333-500 | |
| Sodium Chloride (NaCl) | Sigma Aldrich | S5150-1L | |
| Sodium dodecyl sulfate (SDS) | ThermoFisher Scientific | BP166-500 | SDS is poisonous if inhaled; handle with care in well ventilated spaces using gloves, eye protection, and an N95-grade respirator when handling |
| Sodium Hydroxide (NaOH) | Fisher Scientific | S318-1 | NaOH is an eye/skin irritant as a solid and corrosive in solution. Handle in a chemical fume hood using gloves, a lab coat, and goggles |
| Spermidine | Sigma Aldrich | S2626 | |
| Standard Inverted Light Microscope | Leica | 11526213 | |
| Standard lab agarose gel materials | Varies | ||
| Standard lab materials such as serological pipettes and pipette tips | Varies | ||
| T4 DNA Ligase | New England Biolabs | M0202S | |
| T4 DNA Polymerase | New England Biolabs | M0203S | |
| T4 PNK | New England Biolabs | M0201S | |
| Tabletop vortexer | Fisher Scientific | 2215414 | |
| Taq DNA Polymerase | New England Biolabs | M0273S | |
| Thermomixer | Eppendorf | 5384000020 | Alternatively, can use a waterbath |
| Tris base | Fisher Scientific | BP152-5 | |
| Triton X-100 | Sigma Aldrich | 9002-93-1 | Triton X-100 is hazardous; use a lab coat, gloves, and goggles when handling |
| Trypsin | Fisher Scientific | MT25052 | |
| Tube rotator | VWR | 10136084 | |
| USER enzyme | New England Biolabs | M5505S |
Referenzen
- Gilmour, D. S., Lis, J. T. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Molecular and Cellular Biology. 5 (8), 2009-2018 (1985).
- Solomon, M. J., Varshavsky, A. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proceedings of the National Academy of Sciences of the United States of America. 82 (19), 6470-6474 (1985).
- Irvine, R. A., Lin, I. G., Hsieh, C. -. L. DNA methylation has a local effect on transcription and histone acetylation. Molecular and Cellular Biology. 22 (19), 6689-6696 (2002).
- Albert, I., et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 446 (7135), 572-576 (2007).
- Blat, Y., Kleckner, N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell. 98 (2), 249-259 (1999).
- Ren, B., et al. Genome-wide location and function of DNA binding proteins. Science. 290 (5500), 2306-2309 (2000).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- He, Q., Johnston, J., Zeitlinger, J. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nature Biotechnology. 33 (4), 395-401 (2015).
- O’Neill, L. P., Turner, B. M. Immunoprecipitation of native chromatin: NChIP. Methods. 31 (1), 76-82 (2003).
- Schmidl, C., Rendeiro, A. F., Sheffield, N. C., Bock, C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nature Methods. 12 (10), 963-965 (2015).
- Cao, Z., Chen, C., He, B., Tan, K., Lu, C. A microfluidic device for epigenomic profiling using 100 cells. Nature Methods. 12 (10), 959-962 (2015).
- Shankaranarayanan, P., et al. Single-tube linear DNA amplification (LinDA) for robust ChIP-seq. Nature Methods. 8 (7), 565-567 (2011).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Grosselin, K., et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nature Genetics. 51 (6), 1060-1066 (2019).
- Adli, M., Bernstein, B. E. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nature Protocols. 6 (10), 1656-1668 (2011).
- Zwart, W., et al. A carrier-assisted ChIP-seq method for estrogen receptor-chromatin interactions from breast cancer core needle biopsy samples. BMC Genomics. 14, 232 (2013).
- Valensisi, C., Liao, J. L., Andrus, C., Battle, S. L., Hawkins, R. D. cChIP-seq: a robust small-scale method for investigation of histone modifications. BMC Genomics. 16, 1083 (2015).
- Brind’Amour, J., et al. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nature Communications. 6, 6033 (2015).
- Schmid, M., Durussel, T., Laemmli, U. K. C. h. I. C. ChlC and ChEC; genomic mapping of chromatin proteins. Molecular Cell. 16 (1), 147-157 (2004).
- Zentner, G. E., Kasinathan, S., Xin, B., Rohs, R., Henikoff, S. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nature Communications. 6, 8733 (2015).
- Skene, P. J., Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 6, 21856 (2017).
- Meers, M. P., Bryson, T. D., Henikoff, J. G., Henikoff, S. Improved CUT&RUN chromatin profiling tools. eLife. 8, 46314 (2019).
- Hainer, S. J., Bošković, A., McCannell, K. N., Rando, O. J., Fazzio, T. G. Profiling of pluripotency factors in single cells and early embryos. Cell. 177 (5), 1319-1329 (2019).
- Kaya-Okur, H. S., et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications. 10 (1), 1930 (2019).
- Carter, B., et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nature Communications. 10 (1), 3747 (2019).
- Gopalan, S., Wang, Y., Harper, N. W., Garber, M., Fazzio, T. G. Simultaneous profiling of multiple chromatin proteins in the same cells. Molecular Cell. 81 (22), 4736-4746 (2021).
- Patty, B. J., Hainer, S. J. Transcription factor chromatin profiling genome-wide using uliCUT&RUN in single cells and individual blastocysts. Nature Protocols. 16 (5), 2633-2666 (2021).
- Zheng, X. -. Y., Gehring, M. Low-input chromatin profiling in Arabidopsis endosperm using CUT&RUN. Plant Reproduction. 32 (1), 63-75 (2019).
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 10 (12), 1213-1218 (2013).
- Buenrostro, J. D., et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 523 (7561), 486-490 (2015).
