A cartoon depicting the SiMPull process is shown in Figure 1A. Coverslips are functionalized using NeutrAvidin as an anchor for biotinylated anti-EGFR antibodies to capture EGFR-GFP from total protein lysates. After washing away unbound protein, the phosphorylated receptors are labeled with an anti-phosphotyrosine (anti-PY) antibody15. Figure 1B shows an image of the hydrophobic array, where multiple samples can be prepared and imaged on the same coverslip. One advantage of this sample holder is that minimal sample volumes of ~10 µL are required. The coverslip can be imaged by placing it directly on the microscope stage. However, it is helpful to stabilize the coverslip by using a coverslip holder. The coverslip holder shown in Figure 1B was created using a 3D printer, and the blueprint is provided in Supplementary Coding File 5. The autofluorescence of the hydrophobic ink is a useful guide to finding the focal plane of the sample (Figure 1C). An example of a multichannel raw image is shown in Figure 1D. An overlay of the raw green and far-red channels is shown in Figure 1E.
Figure 2 outlines the analysis workflow and provides representative data. Data acquisition first starts with acquiring fiducials for channel registration, which is used to overlay the individual spectral channels data (Figure 2A). Bright-field images are taken using a nanogrid pattern that passes white light and is detected in each spectral channel of the image splitter (not shown). The green channel acts as the reference channel, and the far-red channel is the shifted channel. The local weighted mean transform is calculated using the fitgeotrans20 function in MATLAB and is used to shift far-red coordinates into the coordinate frame of the green channel. This transform uses a second-order polynomial model at every control point. Multichannel data of the SiMPull array is then acquired. This workflow consisted of a semi-automated acquisition, where a starting ROI was selected for the specific sample square, and three regions around this area were imaged, such that each dataset contains the full quad-view image from three independent ROIs (Figure 1D). In each spectral channel, the emitter candidate locations are found by applying a difference of Gaussian filter to images and identifying local maxima. Subregions (boxes, Figure 2B) are drawn around local maxima, and emitter photon counts are estimated by assuming each subregion contains only one emitter. Subregions holding emitter candidates with photon counts above a minimum value are retained for fitting. A Gaussian point spread function (PSF) fits each emitter candidate within small subregions roughly centered around each emitter. The resulting localizations are thresholded based on their photon count, background, Cramér-Rao lower bound of the fit coordinates, PSF variance (i.e., PSF width), and a p-value describing the goodness of fit of the PSF model. A Gaussian image is created for each spectral channel, with uniform intensity Gaussian blobs placed at the coordinates for each good fit (Figure 2C). Colocalization is visualized by overlaying the Gaussian images from each spectral channel using the transform calculated from the fiducial sample (Figure 2D). It is important to fluorescently label the receptor for identification since there is still non-specific binding of the anti-phosphotyrosine antibodies to the surface when cell lysate is present. The EGFR-GFP (green channel) is used to generate a mask of the receptor locations, and only the AF647-anti-PY signal (far-red channel) within that mask is counted (Figure 2D). Pairs within 1 pixel (106.7 nm pixel size) are considered to be colocalized and saved to a list containing the reference channel coordinates. The percentage of AF647 colocalized with GFP is calculated to determine the fraction of phosphorylated receptors (Figure 2E).
There are several critical steps to ensure good data quality. One such effort is to incubate the coverslip array with NaBH4 as described in the protocol to quench autofluorescence in the green channel. This autofluorescence refers to the non-specific signal due to possible impurities on the glass, containing single or conjugated π bonds21. Such impurities are potentially from the aminosilane and PEG reagents used in the functionalization process or dust from the air, and tend to fluoresce in the green spectral channel. Despite efforts to keep glass stored under nitrogen, these molecules may also be generated through oxidation that occurs in storage. NaBH4 has also been used to reduce fluorescence from impurities on slides and microarrays, including those with silane coating16. Figure 3A shows the reduction in the number of background detections that occur when the piranha etched glass is treated with NaBH4. While NaBH4 reduces background fluorescence dramatically, some emitters are still detected in the green channel. One can correct this by acquiring background images from lysate-free samples (Figure 3D) and subtracting the average number of background localizations from the GFP-containing samples. Fluorescence from impurities was not detected in the far-red channel. If the receptor density is too high, multiple GFP emitters can be found within a single diffraction-limited spot (data not shown). Using step-photobleaching to identify the number of GFPs per spot, we found that a receptor density between 0.04-0.08 proteins/µm2 provided sufficient spacing between single emitters to remove the potential of finding multiple emitters per spot12. The receptor density can be optimized by varying the amount of IP antibody bound to the glass surface or the amount of lysate added. It is critical to ensure that the antibody targeting the POI is used at saturating levels. It is recommended to acquire an antibody concentration curve on phosphorylated samples to determine the appropriate labeling conditions (Figure 3B). In addition, the phospho-specificity of an antibody needs to be validated with resting samples and/or treatment with protein-specific kinase inhibitors (Figure 3B). Antibodies will dissociate from the receptor during the imaging time window. Treating the sample with a combination of PFA and GA prevented signal loss (Figure 3C).
Finally, it is important to optimize the single-molecule fitting parameters. The first "box finding" step that identifies potential emitter candidates (Figure 2B) needs to be generous to allow many candidates to undergo the Gaussian Fitting. Thus, the minimum photon threshold for box finding can be relatively low to capture all real emitters and some background spots. It is also important to not set the box size and overlap allowance too small. Keeping the box size within 5-7 pixels and allowing two-pixel overlap is ideal for emitters at the recommended density. After box finding, the minimum photons threshold in the fitting step needs to be optimized. The minimum photons parameter contributes to determining which Gaussian fitted emitters pass as a true fit. To determine the proper minimum photon threshold for true GFP fits, the code includes a histogram plotting function to examine the photons/localization in both background (no cell lysate) and GFP-containing (plus cell lysate) samples (Figure 3D). This step is important because, while NaBH4 reduces the amount of fluorescence from impurities, it does not remove all background localizations. Figure 3D demonstrates the need to set a minimum photon threshold to reduce the number of detections from impurities. To determine this threshold, a histogram of background emitter intensities is calculated from imaging a sample that is not exposed to cell lysate (Figure 3D, top left). The majority of the background emitters were found to have values less than 475 photons. In comparison, the sample containing true GFP emitters showed a significant fraction of the distribution above 475 (Figure 3D, top right). The threshold is chosen by inspection to remove as many background counts as possible while minimizing the amount of signal loss from the lysate sample (Figure 3D, bottom row). The remaining background count density at this threshold is accounted for in the quantitative analysis.
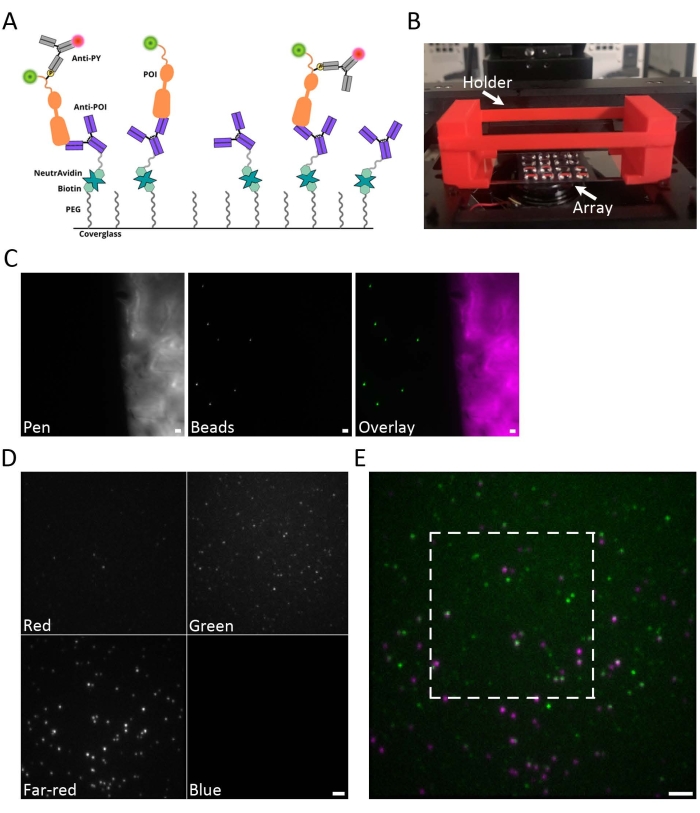
Figure 1: Overview of sample preparation. (A) Cartoon depicting the SiMPull approach. Coverslips are functionalized with an antibody that recognizes the POI to capture that POI from whole cell lysates. The glass is first coated with PEG and biotin-PEG. NeutrAvidin is then bound to the biotin-PEG and acts as an anchor for the biotinylated anti-POI antibody. Phosphorylated proteins are then detected with a fluorescently labeled anti-PY antibody. (B) Photograph of the coverslip holder (red) with coverslip array in place and mounted on the microscope stage. The multisample arrays are generated using hydrophobic ink to create up to 20 individual sample squares on a single glass coverslip. The coverslip is 60 mm x 24 mm. (C) Example images of the hydrophobic ink autofluorescence (magenta) and fluorescent beads (green). The autofluorescence of the hydrophobic ink is a useful guide to find the focal plane at the coverslip surface. (D) Example of a raw data image with spectral channels separated on the camera chip by the Quad-view image splitter. The Quad-view filter set includes the following emission filters: blue (445/45 nm), green (525/45 nm), red (600/37 nm), far-red (685/40 nm). (E) Raw overlay of green and far-red channels. The white box indicates the region further examined in Figure 2B–D. Scale bar (C-E) = 2 µm. Please click here to view a larger version of this figure.
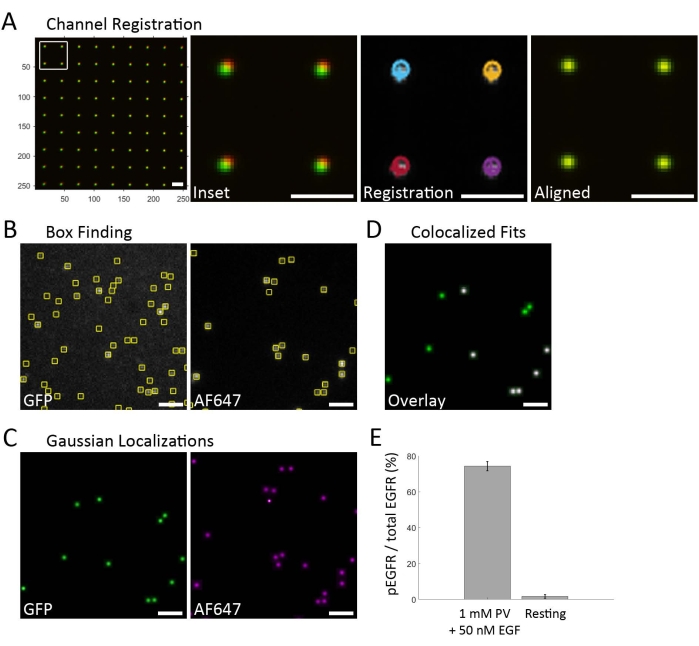
Figure 2: Data analysis workflow. (A) Channel registration is first performed on images acquired from the nanogrid. After cropping the two spectral channels of interest (here, green and far-red), the fiducial images for each channel are overlaid (left). Enlargement of the box in the left image (Inset) shows that the images are not yet truly registered. The emitters in each channel fit a Gaussian model and are localized (Registration). Localization of emitters is shown as circles for the far-red channel and crosses for the green channel. The final step is to apply a local weighted mean transform to shift the far-red channel localization coordinates into the green channel reference frame (Aligned). The calculated local weighted mean transform is then used to register the subsequent SiMPull data. (B) Representative images of the green/EGFR-GFP channel and the far-red/AF647-anti-PY channel. Single emitters above the background photon count are identified and marked with boxes. (C) The emission profile within each selected box is fit to a Gaussian model, and the emitters that fit the model of a single fluorophore PSF are kept. (D) A mask is created from the GFP emitters to identify the location of EGFR-GFP (green). Colocalization of EGFR-GFP and AF647-anti-PY identifies phosphorylated receptors (white). (E) The fraction of phosphorylated receptors is calculated from the colocalized EGFR-GFP and AF647-anti-PY fits. The bar graph compares PV + EGF treatment to resting cells, averaged for multiple measurements. Error bars represent standard error calculated assuming a binomial distribution. Scale bar = 2 µm. Please click here to view a larger version of this figure.
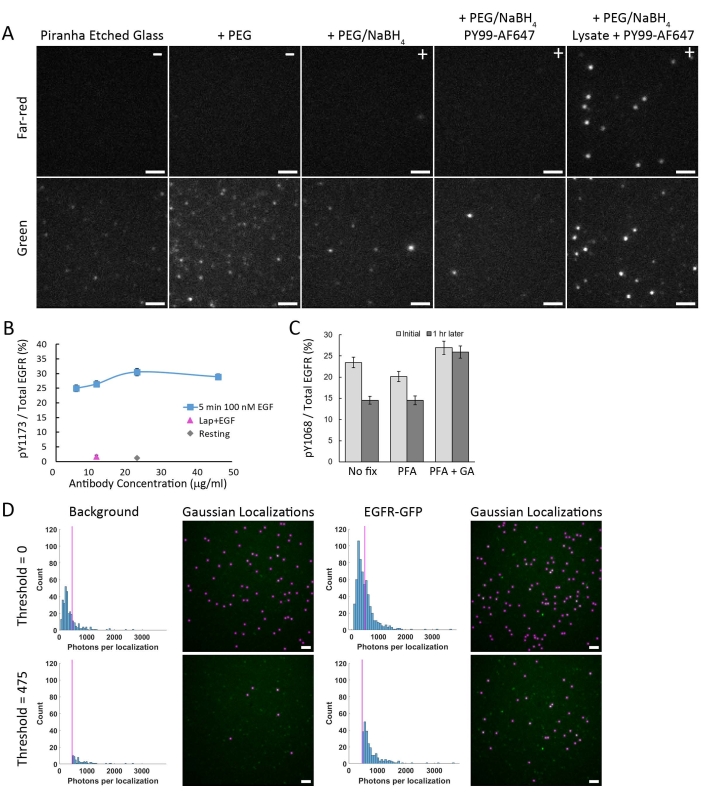
Figure 3: Critical steps to ensure data quality. (A) From left to right, the first three panels are representative images of the autofluorescence on glass under the respective conditions: after piranha etching, with PEG, and PEG plus NaBH4 treatment (indicated with +). Additionally, surface functionalization is retained after NaBH4 treatment as demonstrated by minimal non-specific PY99-AF647 binding while retaining robust binding of EGFR-GFP from the lysate. (B) A saturation curve must be acquired for each batch of antibodies used to ensure optimal antibody labeling. This figure shows the concentration curve for labeling EGFR with the site-specific phosphotyrosine antibody, anti-EGFR-pY1173. Minimal phosphorylation is detected in untreated cells (Resting, gray diamond). As a control for non-specific binding, cells were also treated with the EGFR kinase inhibitor, Lapatinib, before adding 100 nM of EGF (magenta triangle), which shows the expected prevention of EGFR phosphorylation. Error bars represent standard error assuming a binomial distribution. (C) Fixation of the sample with a combination of PFA and GA prevents antibody dissociation over time. Error bars represent standard error assuming a binomial distribution. (D) False positives are excluded by selecting the appropriate threshold for Gaussian fitting. Comparing the histogram of fit intensities at a low threshold (Threshold = 0; top) between background (no lysate) and real data (plus cell lysate) allows for selection of appropriate value (Threshold = 475; bottom) to remove fits from autofluorescent spots in the green channel. The vertical magenta line indicates a 475 photon threshold. Histograms are calculated from the same number of ROIs for each sample type (n = 3). Scale bar = 2 µm. Please click here to view a larger version of this figure.
Supplementary Coding File 1: zip file containing scripts and utilities for running SiMPull analysis. Please click here to download this File.
Supplementary Coding File 2: zip file containing the smite single-molecule analysis package. Please click here to download this File.
Supplementary Coding File 3: zip file containing the sample data. Please click here to download this File.
Supplementary Coding File 4: zip file containing representative sample data analysis outputs. Please click here to download this File.
Supplementary Coding File 5: Coverslip holder blueprint for 3-D printing. Please click here to download this File.
