All procedures were conducted following the policies of the Oswaldo Cruz Foundation and the National Ethical Council (CAAE: 59902816.7.0000.5091). The human protocols were developed in collaboration with the clinical research group from the Research Center for Tropical Medicine of Rondônia (CEPEM), which was in charge of enrolling patients in the study. Informed consent was obtained from all the patients.
For the animal study, procedures were performed following the principles of conduct of the Brazilian Practice Guide for the Care and Use of Animals for Scientific and Didactic Purposes of the National Council for the Control of Animal Experimentation (CONCEA). The protocols were approved by the Fiocruz Animal Experimentation Council (CEUA protocol LW15/20-2).
1. Collection of human blood samples and PBMC isolation
- Collect blood of Plasmodium-infected patients in a 10 mL evacuated blood collection tube with sodium heparin. As malaria patients display lymphopenia, there is a range of 5-9 x 106 peripheral blood mononuclear cells (PBMCs) in a 10 mL blood sample. Since CD8+ T cells or γδ T cells make up ~10% of PBMCs, preferably collect 50-100 mL of blood/patient.
NOTE: A minimum of 1 x 105 effector cells/ condition should be considered. - Calculate the percentage of iRBCs by blood smear as described below.
- Add 5 μL of total blood to a clear slide and prepare a blood smear. Perform Romanowsky-type panoptic fast stain or May-Günwald-Giemsa stain. Here, the panoptic fast stain kit is used, which is composed of three reagents: reagent A (fixation), reagent B (cytoplasmic stain), and reagent C (nuclear and cytoplasmic differential stain).
- Slowly dip the slide in solution A 10x, then 4x in solution B, and finally 10x in solution C. Drain out any excess reagent from the slides between solutions. After dipping in solution C, rinse the slide in running tap water and let it dry.
- Under an upright light microscope with a 100x oil immersion objective, count 1000 RBCs in sequential squares and calculate the percentage of parasitemia using the following equation:
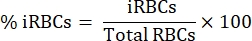
- Dilute 15 mL of blood in a 1:1 proportion in sterile phosphate-buffered saline (PBS).
- Add 15 mL of lymphocyte separation medium (density of 1.077g/mL) to a 50 mL tube. Carefully layer the 30 mL of diluted blood sample onto the centrifugation medium solution. When layering the sample, do not let the blood sample and lymphocyte separation medium mix.
- Centrifuge the tubes at 400 x g for 40 min at 22 °C, with low acceleration and no break setting.
- Draw off the upper layer containing plasma using a sterile pipette, leaving the mononuclear cell layer undisturbed. Transfer the layer of mononuclear cells (PBMCs) to a sterile tube using a sterile pipette.
- Do not discard the tube containing the blood pellet as it will be used later for infected RBC isolation. From this step, be sure to keep the RBCs at room temperature (RT). Never let them cool down.
- Wash the cells twice by adding PBS and centrifuge at 350 x g for 10 min, with break setting. Resuspend the cell pellet in 5 mL of RPMI medium supplemented with penicillin/streptomycin and 10% fetal bovine serum (FBS; complete medium).
- Count the PBMCs in the presence of trypan blue solution to check cell viability, using a hemacytometer (Neubauer chamber) or automated cell counter. Do not count the blue-stained cells as these represent dying cells, which absorb trypan blue.
- Adjust the cell concentration to 107 cells/mL using complete medium. Purify the desired cytotoxic lymphocyte populations (CD8+ T cells, NK cells, γδ T cells) using magnetic beads isolation as per reagent manufacturer protocol.
2. Human RBC isolation
NOTE: For human-infected RBC isolation, it is recommended to start with blood samples that have a minimum of 2% parasitemia, preferentially with more in the trophozoite/early schizont parasite stage.
- Prepare the PERCOLL (from hereafter referred to as density gradient separation medium) at the recommended concentration as described below.
- Add 90 mL of 100% density gradient separation medium and 10 mL of 10x PBS to obtain 90% isotonic density gradient separation medium.
- For P. vivax-infected reticulocyte separation, prepare45% density gradient separation media. Add 50 mL of 90% isotonic density gradient separation medium and 50 mL of 1x PBS to obtain 45% density gradient separation medium.
- For P. falciparum-infected erythrocyte separation, prepare 65% density gradient separation media. Add 72 mL of 90% isotonic density gradient separation medium and 28 mL of 1x PBS to obtain 65% density gradient separation medium.
- For uninfected reticulocyte separation, prepare 70% density gradient separation media. Add 78 mL of 90% isotonic density gradient separation medium and 22 mL of 1x PBS to obtain 70% density gradient separation medium.
- Warm the density gradient separation medium to 37 °C in a water bath.
- After removal of the PBMC layer, carefully remove as much of the top layer of centrifugation medium as possible without touching the cells. With a glass Pasteur pipette, carefully remove the upper neutrophil layer without disturbing the RBC pellet and estimate pellet volume. Add 4x the RBC pellet volume of RT complete medium and resuspend.
- In a second 50 mL conical tube, add 5x the pellet volume of 45%, 65%, or 70% density gradient separation medium depending on the cell type to isolate. Layer the RBC suspension on top of the density gradient separation medium layer carefully.
- Spin for 15 min at 850 x g, with low acceleration and no break setting. Collect the cloudy red/brown layer lying in between the supernatant and density gradient separation medium with a 5 mL pipette. Transfer to a sterile 15 mL tube.
- Wash the cells by adding RT complete medium up to 15 mL and spin down for 10 min at 860 x g. Repeat the washing by adding 10 mL of RT complete medium and spin down. Discard the supernatant and prepare a blood smear from the pellet to check for iRBC enrichment, following step 1.2.
- Resuspend the pellet in 1 mL of RT complete medium and let sit at room temperature while counting the cells. Add 10 μL of cell suspension in a hemocytometer (Neubauer chamber) and count the RBCs in the central area subdivided into 25 medium squares. Adjust cell concentration to 1 x 107 iRBCs/mL in RT complete media.
3. Experimental malaria infection in mouse
- Thaw an aliquot of cryopreserved P. yoelii 17XNL:PyGFP (MRA-817), a GFP-expressing strain obtained from MR4/ATCC, and inject 100 μL intraperitoneally (i.p.) in an 8-week-old female C57BL/6 mouse.
- Follow the parasitemia burden every 3 days by tail vein lancing blood collection.
- Puncture the vessel with the needle bevel up, entering the vein at a shallow angle beginning at the distal end of the tail.
- Collect the blood sample with a pipette or capillary tube up to 5 or 10 mL, then apply manual pressure to stop the bleeding.
- Prepare a blood smear (as described in step 1.2) until parasitemia reaches 10%-15% of infected RBCs.
- Collect 10 μL of blood by tail vein lancing and dilute to 100 μL of PBS to i.p. inject into the second donor mouse.
NOTE: Cryopreserved parasites should be thawed and passed in mice twice before being used for experimental infections. - When the second passage reaches 15% of parasitemia, collect five drops of blood by the tail clipping method and dilute in 1 mL of PBS. Prepare an infection solution by adjusting the concentration to 1 x 106 iRBCs/mL in sterile PBS and inject i.p. 100 μL (1 x 105 iRBCs) of the solution into each mouse required for the experiment.
- Monitor parasitemia every 2-3 days until it reaches ~30% iRBCs, which occurs approximately 12 days post-infection. When the mice reach the desired parasitemia, collect blood by cardiac puncture as described below.
- Aspirate 100 µL of heparin solution (30 U/mL) into a 1 mL syringe with a 26 G needle.
- Anesthetize the mouse by inhalation with 5% isoflurane and confirm the absence of reflexes. Place the mouse on its side and perpendicularly insert the needle just below the elbow, through the ribs, and into the heart. Pull out the syringe plunger slowly and rotate the needle until 0.5-1 mL of blood is obtained.
- Perform humane euthanasia by cervical dislocation under anesthesia. Aseptically clean the left side of the mouse with 70% ethanol. With surgical scissors, make a cut on the left side of the mouse passing through the skin and peritoneum. Locate the spleen and remove it.
- Place the mouse spleen in a Petri dish with 5 mL of complete medium to purify the desired effector cell population (e.g., CD8+ T cells).
4. Obtaining fresh whole mouse splenocytes
- In the Petri dish, carefully cut the spleen into small pieces using scissors or a razor.
- Place a 100 µM cell strainer over a 50 mL conical tube and transfer the excised spleen into the cell strainer with a pipette. Mash the spleen through the strainer with a syringe plunger.
- Wash the cells through the strainer with 10 mL of complete media. Centrifuge the cells at 300 x g for 10 min at 4 °C, then discard the supernatant.
- Resuspend the cell pellet in 2 mL of cold 1x RBC lysis buffer. Incubate the suspension for 5 min on ice.
- Wash the cell suspension with 10 mL of 4 °C complete media. Repeat the wash step three times and remove any cell clots between washes for spleens from Plasmodium-infected mice.
- Centrifuge the cells at 400 x g for 5 min at 4 °C, then discard the supernatant.
NOTE: Plasmodium-infected spleens are enlarged and filled with phagocytic cells containing degraded hemoglobin and hemozoin. - To avoid any issues during the magnetic bead isolation of effector cells, use the following steps to remove hemozoin-enriched phagocytic cells and hemozoin. Resuspend the splenic cells with MACS buffer and adjust concentration to 1 x 108 cells/mL. Place an LS or LD column in the magnetic field. Prepare the column by rinsing with 3 mL of MACS buffer.
- Apply cell suspension onto the column. Wash the column three times with 3 mL of MACS buffer. Collect flow-through containing the splenic cells.
- Count the splenocytes in a trypan blue solution and check cell viability in a hemacytometer (Neubauer chamber) or automated cell counter. Adjust cell concentration to 1 x 107 cells/mL in RT complete media.
5. Cytotoxic effector cell purification (CD8a+ T cell negative selection)
NOTE: There are many positive and negative selection reagents that can purify cytotoxic effector cells (CD8+ T, γδ T, NK, iNKT, MAIT cells). In this protocol, we use a negative selection of splenic CD8a+ T cells and follow manufacturer instructions.
- Centrifuge all splenocytes at 400 x g for 10 min at 4 °C and discard the supernatant. Resuspend the cell pellet in 40 µL of MACS buffer.
- Add 10 µL of a biotin-antibody cocktail. Mix well and incubate for 5 min on ice.
- Add 30 µL of MACS buffer. Add 20 µL of anti-biotin micro-beads. Mix well and incubate for 10 min on ice.
- Add 400 µL of MACS buffer and proceed to magnetic cell separation. Place the MACS LS column in the magnetic field support. Prepare the column by rinsing with 3 mL of MACS buffer.
- Apply cell suspension onto the column. Wash the column three times with 3 mL of MACS buffer. Collect the flow-through containing all unlabeled cells, which are the enriched CD8a+ T cells.
- Count the CD8a+ T cells in a trypan blue solution and check cell viability in a hemacytometer (Neubauer chamber) or automated cell counter. Adjust the cell concentration to 1 x 107 cells/mL in RT complete media.
6. P. yoelii infected RBC isolation
- Centrifuge the collected blood in a 1.5 mL tube at 850 x g for 3 min. Discard the serum and resuspend the blood in 1 mL of 1x RPMI without FBS.
- Place the LS column in the magnetic field support and rinse with 3 mL of 1x RPMI. Pass the RBC suspension through the column. To isolate more iRBCs, reapply the flowthrough (3 mL of RPMI and 1 mL of diluted blood) into the column.
- Wash twice with 5 mL of 1x RPMI. Perform washing steps by adding buffer aliquots as soon as the column reservoir is empty. Do not let any columns dry up.
- Add 5 mL of RPMI, remove the column, and purge the iRBCs into a new 15 mL tube. Count the iRBCs and adjust the concentration to 1 x 107 RBCs/mL.
7. CFSE labeling of RBCs and preparation for flow cytometry
NOTE: Start the RBC labeling protocol with twice the number of cells that will be used for the experiment, since ~50% of the cells are typically lost in the washing steps following the CFSE labeling step. To establish the autofluorescence of the RBCs/iRBCs, include a control sample of unlabeled cells.
- Dilute CFSE to a final concentration of 10 mM in 1x RPMI without FBS. Wash the cells once with 1x RPMI without FBS in a 15 mL tube.
- Resuspend the RBCs in 500 μL of 1x RPMI without FBS and add 500 μL of the diluted CFSE. Incubate for 8 min at RT, protected from light.
- Wash three times by adding 14 mL of complete medium (10% FBS RPMI), followed by centrifugation at 850 x g for 10 min. Resuspend the cells in complete medium to a concentration of 1-5 x 106/ mL. Incubate for 1 h at RT.
8. Cytotoxic lymphocyte/RBC coculture and preparation for flow cytometry
NOTE: If the involvement of specific receptors or molecules will be measured, incubate the specific blocking and isotype control antibodies (10 mg/mL) with the effector cells for 30 min before coculture.
- Plate the cells in a 96-well round-bottom plate. Add the purified lymphocytes and CFSE-labeled iRBCs in the desired effector-target cell ratio to a final volume of 200 μL and homogenize. Prepare each condition in triplicate.
NOTE: We suggest adjusting the iRBC concentration to 1-5 x 104 cells/mL and choosing the ratio based on the purified cell number (e.g., 0.5:1, 2.5:1, and 5:1). - Include the spontaneous lysis control, which is the target iRBCs without the effector, to evaluate any spontaneous lysis, as this will be used to represent a 100% cell viability condition.
- Spin down the plate for 1 min at 360 x g to maximize cell contact. Incubate at 37 °C and 5% CO2 for 4 h.
NOTE: Hypoxia conditions (low O2), systematically used for P. falciparum culture, should not be used since the effector cells do not survive in this environment. In contrast, Plasmodium parasites are viable in ambient air in 5% CO2 for up to 12 h. - Spin for 5 min at 850 x g and flip the plate to remove the supernatant.
NOTE: If desired, the supernatant can be used to measure any soluble factors released in the coculture. - Label the RBCs with anti-mouse Ter119 1:200 (or anti-human CD235 1:100 for human samples) and CD8+ T cells with anti-CD8 1:200 or anti-CD3 1:200 antibodies (anti-mouse or human) for 30 min at 4 °C in 1x PBS containing 3% FBS (FACS buffer).
NOTE: The antibody fluorophore should be selected based on the cell tracer/cell proliferation reagent (e.g., CFSE-labelled iRBC, APC-Cy7 anti-Ter119, and PerCP-Cy5.5 anti-CD8a). - Wash the cells with FACS buffer and spin down for 5 min at 850 x g. Transfer the samples to FACS tubes and add 30 μL of counting beads to individual tubes. Homogenize the count beads by vortexing for 30 s.
9. Flow cytometry
- Analyze samples using a 405/488/561/640 laser instrument.
- For human cells labeled with CFSE (FITC), PE anti-human γδ TCR, and PE-Cy7 anti-human CD235a antibody, use 530/30 (FITC), 575/25 (PE), and 780/60 (PE-Cy7) filters in a three-laser (Blue, Red, Yellow-Green) configuration cytometer.
- For mouse cells labeled with CFSE (FITC), PerCP-Cy5.5 anti-mouse CD8a, and APC-Cy7 anti-mouse Ter119, use 530/30 (FITC), 695/40 (PerCP-Cy5.5), and 780/60 (APC-Cy7) filters in the three-laser cytometer setup.
- Choose the counting beads population as the stopping gate to acquire a minimum of 20,000 events in the stop gate, which should be identical for all conditions. Perform analysis and compensation using appropriate flow cytometry acquisition/analysis software.
- Set the gating strategy as described below.
- Select single cells (singlets), excluding debris using FSC peak height (H) to area (A) ratio. Select RBCs and exclude the lymphocytes from analysis based on fluorescence of the RBC marker (Ter119 or CD235a) and the lymphocyte-specific antibody (CD8a).
- In the previous RBC gating, select viable RBCs based on CFSE positive staining. Analyze the data with flow cytometer data analysis software.
10. Calculation and statistics
- To calculate the percentage of iRBC lysis, follow the gating strategy described in step 9. The percentage of viable RBCs is the frequency of CFSE-positive cells inside the RBC gate based on Ter119 (mouse) or CD235a (human) positive fluorescence.
- Use the spontaneous lysis control, RBCs without effector cells, condition as a control to estimate RBC spontaneous rupture. This condition will be considered RBC lysis (100% viability). Use the following formula to calculate the percentage of RBC lysis in each tested condition:
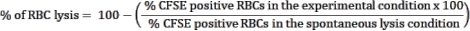
NOTE: During the culture incubation, some infected RBCs may spontaneously lyse or be ruptured by the parasite. Use the CFSE-positive RBC frequency of the spontaneous lysis condition, which contains no effector cells, as the baseline for lymphocyte cytotoxicity. - Calculate the average of triplicates for each condition and determine statistical significance using two-way ANOVA at multiple comparisons.
Here, the applied methodology for isolation of CFSE-labeled Plasmodium-infected RBCs in a coculture assay with cytotoxic lymphocytes is described. First, we provide a schematic representation of how to perform the protocol, employing human samples infected with P. vivax (Figure 1). Then, an illustrated flowchart on how to proceed with the protocol in a malaria experimental model using a C57BL/6 mouse infected with P. yoelii (Figure 2). In Figure 3, the expected results (before and after) for step 6 of the protocol (enrichment of P. yoelii-infected RBCs using magnetic columns) are shown. Finally, Figure 4 shows a representative flow cytometry analysis, detailing the gating strategy needed to assess the percentage of RBC lysis, as described in step 9. Therefore, this section highlights the different techniques used here, describing the entire process of acquisition, assembly, and data analysis to assess iRBC killing by cytotoxic lymphocytes.
Human Plasmodium-infected RBCs lysis by cytotoxic cells
A schematic representation of the protocol to evaluate the killing of human Plasmodium-infected RBCs by cytotoxic cells is shown in Figure 1. To measure cell lysis, PBMC from Plasmodium-infected patients was purified using the separation medium, followed by magnetic purification of the cytotoxic lymphocyte (CTL) population of interest (e.g., CD8+ T, γδ T, NK, iNKT, MAIT cells, etc.). The RBCs' pellet remaining from the PBMC isolation was used to enrich Plasmodium-infected RBCs using density gradient separation medium (65% P. falciparum; 45% P. vivax). iRBCs were labeled with CFSE and incubated with or without lymphocytes for 4 h at 37 °C, 5% CO2. After the coculture period, all RBCs (infected or not) were labeled with anti-human CD235a antibody (for the RBCs) and CTL-specific antibodies (e.g., anti-CD8, anti- γδTCR, etc.) before flow cytometry analysis. The acquisition and analysis of human samples were similar to those demonstrated for mouse samples in Figure 4.
P. yoelii-infected RBC lysis by cytotoxic CD8+ T cells
A flow chart of the experimental procedure to evaluate the cytotoxic effector mechanism in the experimental malaria model is shown in Figure 2, along with representative experimental data shown in Figure 3 and Figure 4. C57BL/6 mice were infected with 1 x 105 P. yoelii (Py) iRBCs, and parasitemia was monitored until it reached around 30%. Py-iRBCs were isolated from blood using magnetic separation, as the parasite heme subproduct, hemozoin, is iron-enriched and functions as paramagnetic particles in a magnetic field13. The purity of the enriched sample was analyzed by blood smear in comparison with the pre-column sample (Figure 3). iRBCs were then labeled with the cell tracer reagent CFSE. In parallel, cytotoxic CD8+ T cells were purified from splenocytes using a magnetic negative selection kit. As a control, the same protocol was used for CD8+ T cell purification from uninfected mice. Effector (CD8+ T) and target (CFSE-labelled Py-iRBC) cells were incubated at different effector:target ratios (E:T: 0.5:1, 2.5:1, and 5:1) for 4 h at 37 °C 5% CO2. At the end of the coculture, each condition was labeled with antibodies anti-mouse Ter119 for RBCs and anti-CD8a for cytotoxic cells. The gating strategy and sample results are represented in Figure 4.
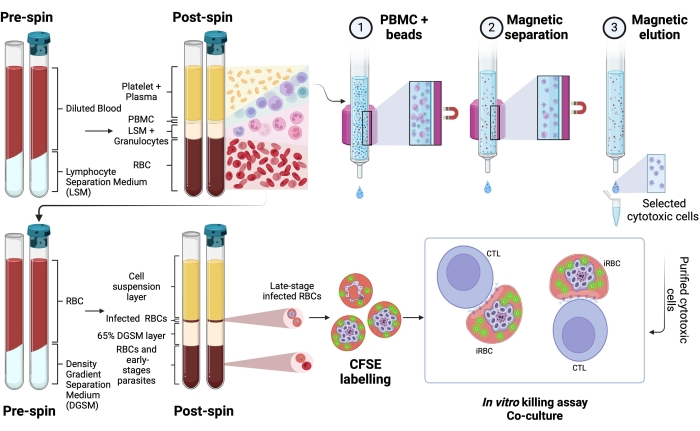
Figure 1. Schematic of the in vitro killing assay. Example of killing assay experiment. Plasmodium-infected blood is collected and processed using a separation gradient medium. After centrifugation, the PBMC layer is collected, and cytotoxic cells (CTLs) were purified using magnetic beads. The RBC layer is once again processed using density gradient separation media. After centrifugation, the Plasmodium-iRBC layer is collected and labeled with CFSE. Thereafter, the purified CTL and iRBCs were cocultured for 4 h at 37 °C, 5% CO2, followed by cell-specific labeling and analysis in a flow cytometer. Please click here to view a larger version of this figure.
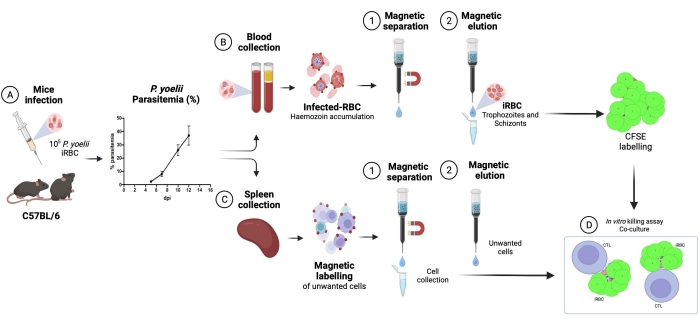
Figure 2. Experimental malaria model for killing assay. First, C57BL/6 mice were infected with 105 PyX17NL-infected red blood cells. Then, parasitemia is monitored by blood smear, until the peak of infection (30%-40% iRBCs), approximately 12 days post-infection (dpi). On the next day, mice were bled and euthanized for spleen collection. The blood was processed, and infected RBCs were enriched by magnetic separation, followed by labeling with CFSE (upper right). The spleen was processed for cytotoxic lymphocytes isolation, by negative selection using magnetic beads. Afterward, the purified cytotoxic lymphocytes and iRBCs were cocultured for 4 h at 37 °C, 5% CO2, followed by cell-specific labeling and analysis in a flow cytometer. Please click here to view a larger version of this figure.
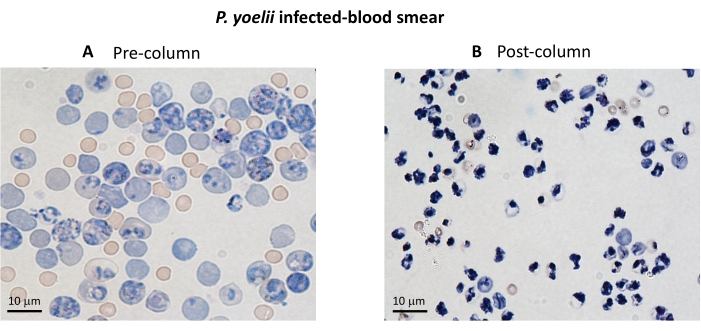
Figure 3. iRBC enrichment using magnetic columns. The iRBCs were purified from P. yoelii infected mice using LS columns. The purification is highlighted by blood smears from blood collected before (A) and after (B) column enrichment. Scale bar = 10 µm. Please click here to view a larger version of this figure.
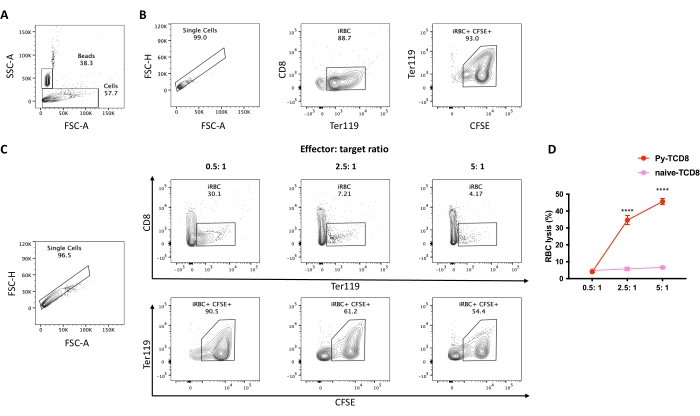
Figure 4. Assessment of cell lysis percentage of P. yoelii-iRBCs by CD8+ lymphocytes. The gating strategy for cell lysis percentage. (A) Set the stopping gate into the counting beads population (~20,000 events). (B) Control spontaneous lysis, iRBCs without lymphocytes. Start the gating strategy by selecting the single cells, excluding debris, based on the FSC peak height to area ratio, followed by the selection of the RBC population in APC-Cy7 Ter119 positive and PerCP-Cy5.5 CD8 negative. Then select live iRBCs as CFSE (FITC) high positive. (C) Example experiment analysis using different effector: target ratios, displaying the percentage of live RBC after the coculture. (D) Graphical representation of the RBC lysis percentage calculated based on the formula described in section 10. The significance comparison between the groups (cytotoxic CD8 or naïve CD8) was assessed by two-way ANOVA with multiple comparisons. P < 0.0001 (****). Please click here to view a larger version of this figure.
