The overall workflow of the protocol is described in Figure 1.
1. Preparation of the sample chamber
- Slide and coverslip cleaning
NOTE: These steps aim to clean the surfaces of the slides as well as the coverslips and prepare them for aminosilanization. One critical requirement for conducting single-molecule fluorescence experiments on surface-tethered molecules is a passivated surface. The most reliable and reproducible passivation technique involves covalently attaching inert polymer chains to the glass surface as a dense layer. Polyethylene glycol (PEG) is the most efficient polymer used for surface passivation16. The details of the passivation procedure using PEG (PEGylation) are described below:- Mark holes to be drilled on the slides with a marker (~6 mm apart and away from the edge). Use a Dremel to drill small holes (1 mm diameter) on the glass slide. Immerse the slides in water during the drilling process.
- Wash the slides with acetone to remove residual ink from the marker.
- Remove from acetone and rinse the slides with water, then microwave them for 5 min in water at high power (700 W).
- Clean the slides with water before placing them in a glass staining jar to be sonicated. Place the coverslips in a different staining jar.
- Sonicate the slides and coverslips in acetone for 30 min in a bath sonicator at 23 °C.
- In the meantime, clean a glass flask for preparing the aminosilanization solution used during the next step. Fill the flask with 1 M KOH, sonicate the flask for 30 min, thoroughly rinse out the KOH with water, and then sonicate for an additional 30 min in methanol. Leave the flask in methanol until the time of the aminosilanization step.
- In the meantime, remove the aminosilane, mPEG, and biotin-PEG from the freezer (−20 °C) and allow them to come to room temperature (RT) in the dark.
- Dispose of acetone from the slide and coverslip jars in the appropriate chemical waste container, rinse thoroughly with water, and then sonicate the slides in 5 M KOH for 30 min.
- Rinse the slides and coverslips with water, and then sonicate in methanol for 2 min (repeat twice). Leave the jars filled with methanol until the aminosilanization step.
NOTE: In this study, deionized water is used unless otherwise mentioned. A bath sonicator working at 23 °C is used.
- Aminosilanization
NOTE: This step aims to covalently link aminosilane to the clean slide and coverslip surface. Functionalized mPEG and biotin-PEG will covalently bind to this surface in the next step.- Prepare an aminosilanization mixture in a clean flask (step 1.6). Prepare the solution by mixing methanol (150 mL), acetic acid (7.5 mL, use glass pipette), and aminosilane (2.5 mL).
- Mix the solution gently in the chemical fume hood, then pour it into the slide and coverslip jars. Use 50 mL for the coverslips and 100 mL for the slides. Ensure slides and coverslips are fully immersed in the solution.
- Incubate for 10 min, then sonicate the jars for 1 min (sonication removes the impurities from the surface), and incubate for another 10 min.
- Dispose of aminosilanization solution in the waste collection container. Add methanol to the jars, close the jars with lids, and shake gently by hand. Dispose of the methanol, and fill jars with water.
- Return the aminosilane bottle to the freezer (−20 °C).
- PEGylation
NOTE: These steps describe the PEGylation procedure.- During aminosilanization, prepare the pegylation buffer. Weigh 84 mg of sodium bicarbonate (NaHCO3) and add it to 10 mL of water (10 mM). In addition, weigh mPEG and biotin-PEG and set them aside. For six slides and coverslips, use 96 mg of mPEG and 1.2-2.4 mg of biotin-PEG.
NOTE: It is important not to add too much biotin-PEG since it can increase the number of background spots. Do not dissolve the PEG mixture until right before application to slides. - Rinse the slides with water, dry them with a gentle air blow, and then place them in the humidified assembly boxes.
NOTE: It is essential to use a clean airline. Avoid using compressed canned air, which can leave residues on the glass. - Add pegylation buffer to PEG powder mixture and pipette up and down gently multiple times to dissolve. Add 55 µL of pegylation buffer per slide (for six slides, add 330 µL of pegylation buffer).
- Centrifuge at 9,600 x g for 1 min at 23 °C to precipitate undissolved particles. Collect the supernatant to use in the next step.
NOTE: Biotin-PEG hydrolyzes quickly due to the presence of the NHS group. It is important to do the mixing and centrifugation steps quickly. - Apply 60 µL of PEGylation solution to each slide and then place the coverslip on top so that the PEGylation solution is sandwiched between the slide and the coverslip.
NOTE: Avoid introducing bubbles between the slide and coverslip, as this will reduce the passivation efficiency. Remove any bubbles by adjusting the coverslip and the slide with a pipet tip. - Place the slides in a drawer that is flat and dark. Slides can be stored overnight; however, incubation for 4-6 h results in optimum passivation.
NOTE: It is essential to remember which side is pegylated. - Mark the non-pegylated side before storage. After incubation, gently disassemble and rinse the slides and coverslips thoroughly with water
- Dry the slides and coverslips by blowing air. Keep the slides and coverslips in a sterile tube (50 mL), with the PEGylated surface facing away from each other. Store at −20 °C until the day of the experiment.
NOTE: It is best to use slides and coverslips within 4 weeks of preparation. The PEGylated surfaces should face away from each other. Storing the PEGylated slides and coverslips in vacuum-sealed bags can increase their shelf life.
- During aminosilanization, prepare the pegylation buffer. Weigh 84 mg of sodium bicarbonate (NaHCO3) and add it to 10 mL of water (10 mM). In addition, weigh mPEG and biotin-PEG and set them aside. For six slides and coverslips, use 96 mg of mPEG and 1.2-2.4 mg of biotin-PEG.
2. mGluR2 expression with incorporated unnatural amino acid, fluorescent labeling, and extraction
NOTE: This protocol outlines the preparation, reagents, and treatment of cells for expressing mGluR2 containing the UAA 4-azido-L-phenylalanine (AZP). The procedure is for HEK293T cells grown on 18 mm glass coverslips. The procedure can be scaled up as necessary.
- Seeding
NOTE: Maintain the HEK293T cells in DMEM supplemented with 10% (v/v) fetal bovine serum, 100 unit·mL−1 penicillin-streptomycin, and 15 mM HEPES buffer (Supplementary File 1) (pH 7.4) at 37 °C under 5% CO2.- Passage the cells with 0.05% trypsin-EDTA. Seed HEK293T cells on poly-L/D-lysine (PLL/PDL) glass coverslips so that they reach ≥80% confluency during the time of transfection.
- Transfection
- Prepare a 40 mM stock solution of AZP in 0.1 M NaOH.
- Prepare AZP-supplemented media for growing cells and mGluR2 expression. Supplement standard media (+ FBS, pen/strep, 15 mM HEPES) using 40 mM AZP stock solution. Bring the final AZP concentration to 0.6 mM. Add 1 M HEPES solution (half the volume of 40 mM AZP stock solution added). For example, to prepare 10 mL of AZP supplemented media, combine 9.775 mL of standard media, 150 µL of 40 mM AZP stock solution, and 75 µL of 1 M HEPES solution.
- Filter (sterilize) the media using a syringe filter (0.2 µm, PES).
- Replace the standard media with media containing AZP-supplemented media prior to transfection.
NOTE: Be careful not to dry the cells during the replacement of the media. - Transfect the cells with transfection reagent (Table of Materials) following the manufacturer's manual. Transfect HEK293T cells on an 18 mm coverslip using a total of 2 µg of DNA (1000 ng of tRNA/synthetase + 1000 ng of amber codon-containing protein plasmid). Refer to Table 1 for the concentration and volume of components used.
- Change the media 24 h after transfection to fresh AZP-supplemented media and allow the cells to grow for an additional 24 h.
- Labeling with alkyne cyanine dyes
- 20 min before labeling, wash the coverslips with warm (37 °C) recording buffer (RB) (Supplementary File 1) twice, and move them to warm (37 °C) standard media with no AZP (+ FBS, Pen/Strep, 15 mM HEPES).
- Prepare the labeling solution containing Cy3-alkyne, Cy5-alkyne, BTTES, copper (II) sulfate (CuSO4), (+) sodium L-ascorbate, and aminoguanidine.
- Follow the order of making and adding solutions (Table 2):
- Prepare 50 mM BTTES.
- Prepare 100 mM aminoguanidine.
- Prepare 100 mM Na-ascorbate.
- Prepare 655.5 µL of RB.
- Add Cy3/Cy5 alkyne dye (10 mM in DMSO stock) to the RB.
NOTE: Add aminoguanidine to the RB. - Prepare 20 mM CuSO4.
- Mix CuSO4 and BTTES in a new tube (the solution will turn blue).
- Add the CuSO4 and BTTES mixture to RB (2.3.3.6.)
- Add the Na-Ascorbate.
- Follow the volume given in Table 2 below for an 18 mm coverslip.
- Mix the solution thoroughly and incubate on ice and in the dark for 10 min prior to labeling cells.
- Before adding the labeling solution to the coverslips, remove the media and wash them with RB. Add the labeling solution and incubate for 15 min at 37 °C in dark conditions.
- NOTE: To improve labeling, add glutamate (final concentration ~0.5 mM) after 10 min and incubate for an additional 5 min. Copper is very toxic to the cells, and the labeling reaction should not continue for more than 15 min in vivo. Prepare all the components fresh. Add Na-Ascorbate last. Keep the reaction at 4°C while preparing. However, upon the addition of labeling solution, the cells are to be stored at 37 °C inside the incubator.
- Harvesting the cells and extracting the proteins (cell lysis)
- Remove the labeling solution and wash the coverslip (18 mm) containing the mGluR2 transfected cells twice with the RB.
- Using a pipet, wash the cells off the coverslip and resuspend in RB (1 mL).
NOTE: Minimize the sample exposure to light as much as possible after this point. - Pellet the cells by spinning at 1,000 x g at 4 °C for 5 min and remove the supernatant. Resuspend the cell pellet in 80-130 µL of the lysis solution.
NOTE: The cell pellet should be visible by eye. The lysis volume depends on the amount of sample lost during the labeling and washing process. - Mix gently by pipetting to break up the pellet. Wrap in foil and place on the rocker at 4 °C for 0.5-1 h to lyse the cells.
- Pellet the insoluble fraction by centrifugation at 20,000 x g and 4 °C for 20 min. Transfer the supernatant to a fresh cold tube (the lysed protein that contains the fluorescently tagged protein of interest) and store it on ice for experiments.
3. Single-molecule flow chamber assembly and functionalization
- Remove the slide and the coverslip from the freezer and allow them to warm at RT in darkness (~30 min).
- Assemble the chamber using double-sided tape by sandwiching strips of double-sided tape between the slide and coverslip. Make sure the PEGylated surfaces form the interior of the flow chamber.
- Using a pipet tip, press the coverslip to ensure the tape is making complete contact with both coverslip and slide; take care not to break the coverslip. Apply epoxy to the edges of the slides.
NOTE: Do not add so much that epoxy fills in the drilled holes. - Place the flow chamber with the coverslip side facing downward in a humidified dark box to allow the epoxy to dry (~30 min).
NOTE: Add T50 buffer (Supplementary File 1) through the drilled holes to prevent the epoxy from covering the holes (10-15 µL) during the drying period. - Incubate each chamber lane with 500 nM Neutravidin (diluted in T50) by slowly applying ~40 µL to each lane.
- Incubate at RT for 2 min inside a humidified dark box. Wash with ~100 µL of T50 buffer per lane.
- Incubate each chamber lane with 20 nM biotinylated antibody11. The choice of antibody depends on the tag on the protein.
NOTE: If the primary antibody is not biotinylated, first incubate with the biotinylated secondary antibody for 30 min and then incubate with the primary antibody. - Incubate at RT for 30 min inside a humidified dark box. Wash with ~200 µL of T50 per lane.
NOTE: Ensure the lanes never dry out during the preparation process.
4. Single-molecule buffers
- Trolox buffer
NOTE: Trolox buffer is the starting buffer for making imaging buffer. Components of the buffer are dependent on the experiment and may vary depending on the protein of interest. The buffer used in the protocol described here includes salts (NaCl, KCl, CaCl2, MgCl2), buffering agent (HEPES), and Trolox (pH ~7.35).- Dissolve 9-10 mg of Trolox in 10 mL of single-molecule recording buffer (SRB, Supplementary File 1)
NOTE: Trolox makes the buffer slightly acidic. Adjust the pH at this stage using sodium hydroxide (NaOH) solution, 10 M (pH 7.35). The fine pH adjustment will be made after Trolox is fully dissolved; however, increasing the pH at this point increases the solubility of the Trolox. - Mix the solution at RT using a benchtop rocker for 4-8 h (wrapped in aluminum foil) to fully dissolve the Trolox.
- Check the pH and adjust if needed.
- Ensure the Trolox is fully dissolved. Sterilize the solution with a syringe filter and store at 4 °C.
NOTE: The buffer should be used after 2-10 days of aging. Trolox helps to suppress blinking and is commonly used in single-molecule studies17. The anti-blinking properties come from an oxidized derivative of Trolox18; hence, it is recommended to keep it at RT for at least a few hours to mature. Additionally, UV radiation of fresh Trolox solution speeds up the oxidation process and can be used to accelerate the "aging" of Trolox buffer18.
- Dissolve 9-10 mg of Trolox in 10 mL of single-molecule recording buffer (SRB, Supplementary File 1)
- Imaging buffer recipe: Mix Trolox buffer + detergent (~2 times the detergent's CMC value) + 4 mM protocatechuic acid (PCA).
NOTE: The detergent concentration is kept near CMC as high detergent concentrations may result in increased protein denaturation. For example, the following mixture can be used 955 µL Trolox + 5 µL 10% DDM + cholesterol (W%, 10:1) + 40 µL of 100 mM PCA stock solution. Here, PCA acts as an antioxidant agent and was previously used in smFRET studies19. DDM is non-ionic, is commonly used to solubilize membrane proteins20,21, and has been used in single-molecule studies. DDM is a good first choice detergent; however, we recommend testing multiple detergents and ensuring the results are consistent.
5. Microscope setup and smFRET data acquisition
- Turn on the computer and microscope. Turn on the lasers to warm up (532 nm for Cy3 excitation and 640 nm for Cy5 excitation).
NOTE: Here, an inverted microscope equipped with a 100x TIRF objective (N.A. 1.49), image splitter, and EMCCD camera was used. The setup is equipped with a four-line laser combiner, a dichroic mirror, a long pass emission filter, an emission dichroic filter, and a notch filter. - Turn on the EMCCD camera and open the camera software. Wait 20 min for the camera to reach −69 °C and stabilize.
- Mount the sample chamber on the microscope stage. Add the protein sample gradually to achieve ~400 molecules per field of view (step 2.4.5). Wash the chamber with 100 µL of the imaging buffer.
- Adjust the gain, acquisition rate, and laser powers such that single-molecule fluorescence signals are detected in both the donor and acceptor channels. Adjust the concentration of the proteins inside the sample chamber if needed.
NOTE: With more than 400 molecules in the field of view, distinguishing individual molecules becomes more difficult, and the background noise will be higher. - Excite the donor and acquire time traces until at least 80% of the donor molecules in the field of view are photobleached.
- At the end of the movie, turn on the 640 nm laser to directly excite the acceptor until some of the acceptor molecules photobleach, facilitating single-molecule from multimer discrimination.
- Move over to a different field of view and repeat the steps above to collect at least three movies (technical replicates) per condition.
NOTE: Use the lowest laser power possible while selecting a new region of interest (ROI) and focusing to minimize photobleaching. Pay attention to the stage drift during acquisition. If noticeable drift is observed after moving to a new ROI, wait for 3 min before starting acquisition.
6. Data analysis
- Donor and acceptor channel alignment (movie mapping)
- Record the fluorescent bead images in the donor and acceptor channels.
- Generate the mapping file using the bead data to correlate the donor and acceptor fluorescence from each molecule22,23.
NOTE: The emission signal from a single molecule is split into donor and acceptor signals by the emission dichroic filter inside the image splitter. The donor and acceptor images are projected on the camera side-by-side. To accurately associate the donor and acceptor intensities of a single molecule between the two areas, a mapping file is often generated using fluorescent bead samples. Using this mapping file, all the molecules that are detected in the donor and acceptor channels are mapped onto each other. The analysis then generates the time traces, which are the donor and acceptor intensities over time, for each molecule.
- Selection of single-molecule FRET traces (particle picking)
NOTE: Individual particle traces are examined and selected for downstream analysis using MATLAB. The exact selection criteria depend on the system. General guidelines on what constitutes a quality particle are outlined here. All the custom-modified codes are available on GitHub (https://github.com/vafabakhsh-lab).- Select the traces where the total intensity of the traces (donor + acceptor) is stable over time. Select the traces with anti-correlated changes in donor and acceptor intensities.
- Select the donor and acceptor molecules that show single-step photobleaching. Select the traces that are >5 s long.
NOTE: The background in each channel after bleaching should go to zero. The traces should not have many blinking events; this will increase the difficulty of analysis. - Calculate the FRET efficiency using the equation E = (IA− 0.088 × ID)/(ID + [IA− 0.088 × ID])24,25,26, where ID and IA are raw donor and acceptor intensities, respectively.
NOTE: The leakage of donor emission into the acceptor channel is determined using donor-only labeled sample excited using a 532 nm laser26. The leakage correction factor, 0.088, may differ for different setups depending on the filter sets used. It is important to note that quantitative and robust conversion of FRET efficiencies to absolute distances requires the correction of donor and acceptor intensities for multiple factors and has been extensively discussed before27.
- Identify the conformational state by Hidden Markov Modeling (HMM)
- Execute the vbFRET28 program in MATLAB and import the selected traces for a given condition. Set the constraints for the number of potential states and iterations to be executed.
NOTE: Based on the raw data from the representative results, it was hypothesized that there were up to four discrete FRET states occupied by the conformational sensor; thus, a range of one to four states was designated. Improvements in fitting were previously determined to negligibly increase with >25 iterations; thus, 25 iterations were used for fitting the representative data. - Analyze the smFRET traces and export the idealized traces and analysis session. Save the idealized traces to a separate folder for downstream analysis.
NOTE: The programs used to extract state transition and dwell-time data from idealized traces were made available in previously published work29. - Using MATLAB programs, extract state transitions and plot them as a heatmap with the X-coordinate indicating starting conformation and the Y-coordinate indicating ending conformation.
NOTE: Transitions are defined as changes in FRET value >0.1 in the representative results discussed here. The threshold for transitions is dependent on the hypothesized conformational states a protein of interest occupies (set transition threshold to be less than the difference between the closest FRET states), as well as the resolution allowed by the experimental setup. The examination of heatmaps for multiple conditions enables the identification of the most common conformational states a sensor transitions through and, thus, occupies. Four FRET states were identified for the representative results (FRET = 0.31, 0.51, 0.71, and 0.89). - Using MATLAB programs, extract the dwell-times for each identified conformational state. Designate a range of FRET values delineating each state and time resolution during data acquisition. FRET ranges are evenly divided by adjacent FRET states. Export the dwell-times for given treatment conditions.
NOTE: In most cases, dwell-time data can be estimated well by a single exponential decay function. This analysis can be performed in a data analysis and graphing software.
- Execute the vbFRET28 program in MATLAB and import the selected traces for a given condition. Set the constraints for the number of potential states and iterations to be executed.
- Gaussian fitting of smFRET population histograms to quantify state occupancy
- Import population FRET histograms for conditions of interest into the data analysis and graphing software for multiple peak fitting analysis.
- Indicate the number of peaks present (four peaks or states based on HMM analysis). Fitting was performed using multiple Gaussian distributions30 defined as
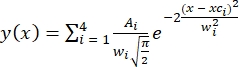 , where A is the peak area, xc is the peak center, and w is the peak width for each peak.
, where A is the peak area, xc is the peak center, and w is the peak width for each peak. - Constrain the fitting parameters as A > 0, xc = FRET ± 0.02, and 0.1 ≤w≤ 0.24. Four FRET peaks for individual population FRET histograms were fit simultaneously. Apply the defined constraints for the fittings of all conditions.
- Calculate the state of occupancy (percentage) as the area of peak of interest divided by the total area, defined as the sum of all peaks.
Expression and fluorescent labeling of UAA-based FRET sensor
Herein, exemplary results of the insertion and fluorescent labeling of a UAA (AZP) within the CRD of mGluR2 (548UAA) are discussed11. As mentioned previously, to insert AZP into mGluR2, co-expression of the engineered translational machinery, which includes a modified tRNA synthetase and complementary tRNA (pIRE4-Azi), and mGluR2 containing an amber codon at position 548, created using mutagenesis, is necessary (Figure 2A,B). The labeling of AZP by cyanine dyes is achieved by a copper-catalyzed cycloaddition reaction (Figure 2C) and results in effective plasma membrane labeling of 548UAA (Figure 2D). To verify the translation of full-length 548UAA and the integrity of the dimeric receptor, SDS-PAGE electrophoresis was performed on the cell lysate from HEK293T cells expressing 548UAA labeled with Cy5. A single band at 250KDa was observed, which coincided with the full-length dimeric mGluR2 (Figure 2E).
Data acquisition and analysis
Cells expressing 548UAA with C-terminal FLAG-tag were labeled with Cy3 (donor) and Cy5 (acceptor) and then lysed with detergent31 in the presence of a protease inhibitor for in vitro study. Upon the completion of cell lysis and removal of the insoluble fraction by centrifugation, the supernatant was applied onto a polyethylene glycol (PEG)-passivated coverslip functionalized with an anti-FLAG-tag antibody for total internal reflection fluorescence (TIRF) imaging (Figure 3). The sample was illuminated using a 532 nm laser, and particles were selected for downstream analysis using smCamera software. Raw donor, acceptor, and FRET traces were generated for all selected molecules in smCamera and selected using MATLAB (section 6; Figure 4). An 8.8% donor bleed-through correction was applied to acceptor intensities for the experimental setup used here. This correction factor will vary with the experimental setup, dichroic filters, and emission filters used and should be determined by measuring the donor and acceptor signals under standard experimental conditions using cells labeled with donor fluorophore only and calculating the bleed-through ([acceptor intensity]/[donor intensity]). Figure 4 shows several representative FRET traces and corresponding donor and acceptor signals. These traces were selected using criteria previously described in the protocol (section 6). Representative selected traces that show transitions between multiple states and donor and acceptor bleaching events are shown in Figure 4B–D.
Identification of conformational states
To identify the conformational states 548UAA occupied and the relationship of these states relative to one another, Hidden Markov modeling (HMM) analysis was conducted. HMM analysis was performed using the vbFRET program executed on MATLAB28 (Figure 5A). Figure 5 uses data from an intermediate glutamate concentration (5µM) to illustrate the process of state identification. Based on raw smFRET traces, it was hypothesized that up to four potential FRET states existed for the CRD. Thus, the number of states was constrained to one to four. Overall, 25 iterations of fitting were performed for each trace to determine the number of states present. From these idealized fits, transitions between discrete FRET states can be extracted and plotted as a transition density heatmap (Figure 5B). The heatmap highlights four discrete FRET states at 0.31, 0.51, 0.71, and 0.89, indicated by dotted lines. Transitions were defined as changes in FRET >0.1. The idealized FRET traces also yield information on dwell-time for each identified conformation (Figure 5C).
Population FRET histogram generation and peak fitting
Representative single-molecule traces for 548UAA in the presence of varying glutamate concentrations that were manually selected for further analysis are shown in Figure 6A,B. A general analysis for smFRET experiments is to generate population histograms from hundreds of smFRET traces for each experimental condition (Figure 6C). Population FRET histograms are created from the segment of traces before bleaching. To avoid biasing the histogram toward the behavior of longer traces, it is necessary to generate a normalized FRET histogram from each trace prior to averaging. This ensures each trace contributes equally to the final histogram. In this system, the FRET histograms show a general shift toward higher FRET with increasing glutamate concentration, indicating a reduction in the distance between the CRDs and a shift toward the active conformation. However, regardless of the glutamate concentration, the FRET signal remains quite sporadic, indicating a high degree of intrinsic dynamics for the CRD. In addition to the general shift toward higher FRET, a redistribution of the conformational ensemble becomes apparent in the histogram (color curves). Individual molecules can also be seen visiting these conformational states (dashed lines) (Figure 6B). To determine the probability of state occupancy, one must divide the area of the peak by the total area, defined as the sum of all four individual peak areas. The smFRET histogram generated from the smFRET experiment of the CRD of mGluR2 displayed four dynamic states with peaks at 0.31, 0.51, 0.71, and 0.89 (labeled at states 1-4), respectively.
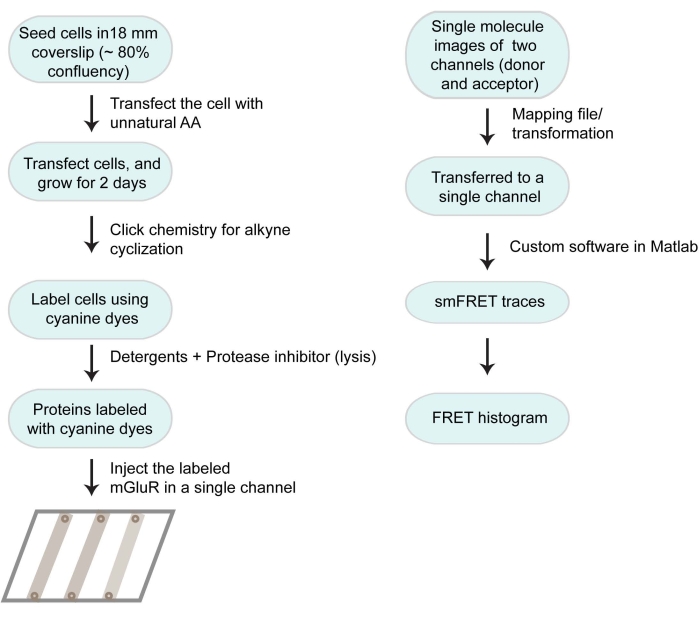
Figure 1: Flow chart of the working protocol and data analysis. Please click here to view a larger version of this figure.
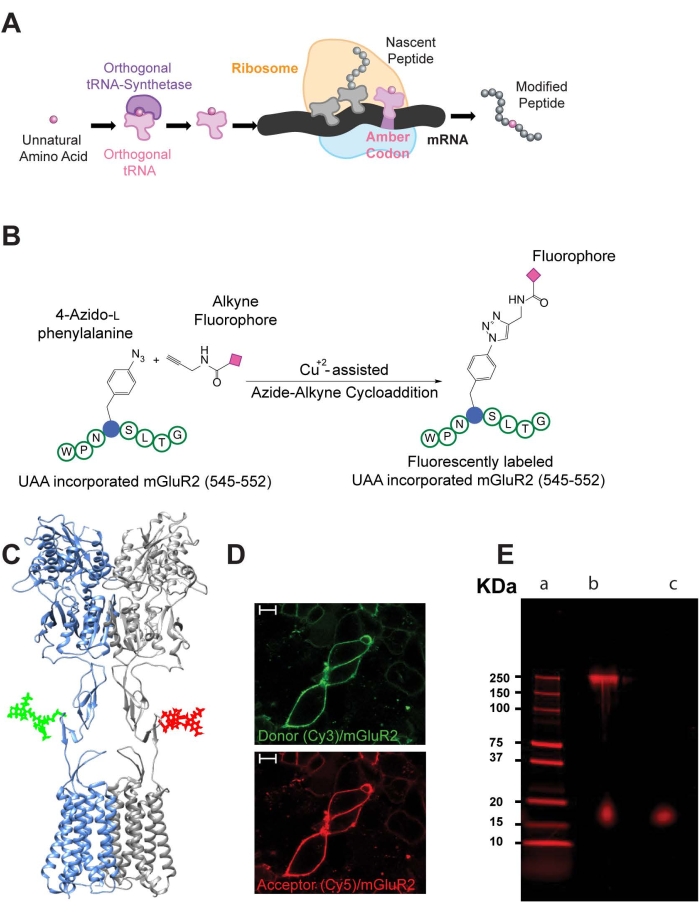
Figure 2: Site-specific labeling of mGluR2 by click chemistry. (A) Schematic of the unnatural amino acid 4-azido-L-phenylalanine incorporation process in cells. (B) Schematic showing site-specific fluorescent labeling of mGluR2, with the unnatural amino acid at position 548, by copper-catalyzed azide-alkyne click reaction. (C) Three-dimensional structure of fluorescently labeled mGluR2 (donor molecule in green, acceptor molecule in red) with Cy3 and Cy5 molecules docked. (D) Representative confocal microscope image of HEK293T cells expressing 548UAA with the cell surface population labeled with donor (green: Cy3) and acceptor (red: Cy5) fluorophores through click chemistry. Scale bars = 10 µm. (E) Image of non-reducing 4%-20% polyacrylamide gel electrophoresis of cell lysate from HEK293T cells expressing 548UAA and labeled by Cy5-alkyne. The gel is imaged with a 633 nm excitation wavelength and 670-BP30 emission filter-Lane a: protein ladder; lane b: cell lysate; lane c: Cy5-alkyne dye. Results are representative of an individual experiment. Panels B, D, and E are reused from Liauw et al.11. Please click here to view a larger version of this figure.
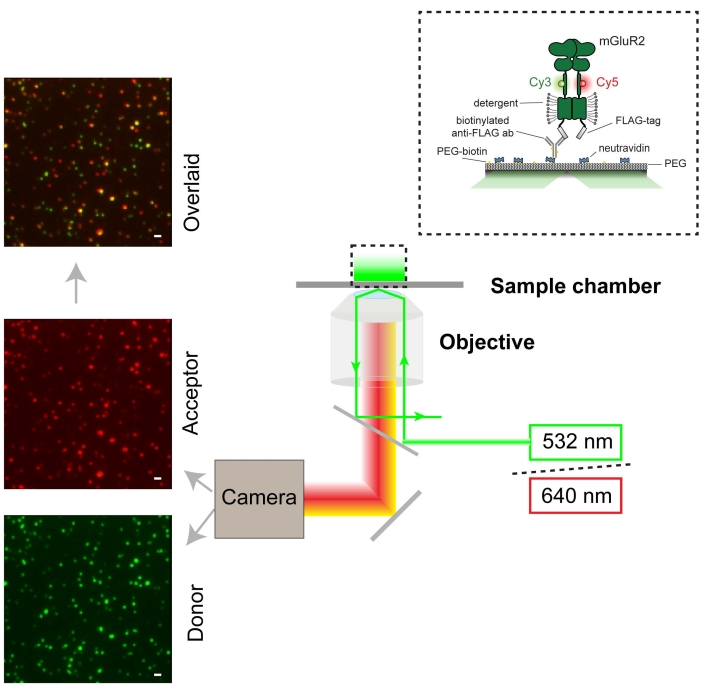
Figure 3: Schematic representation of the smFRET experiments and microscope setup (TIRF). The single-molecule fluorescence image of the donor is shown in green (scale bar = 1000 nm) and the acceptor in red. The donor was excited at 532 nm using a laser. The acceptor is excited by FRET from the donor. Please click here to view a larger version of this figure.
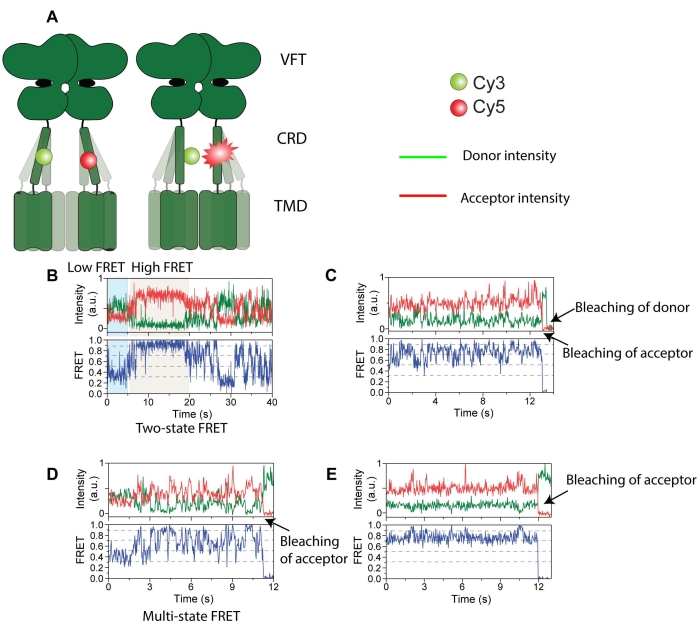
Figure 4: Intensity traces of donor (green) and acceptor (red) labeled mGluR2. (A) Cartoon showing the receptor dynamics and change in distance between FRET probes. (B) Long-lived high FRET state. (C) Multiple FRET states with acceptor photobleaching first followed by donor photobleaching. (D) Short-lived FRET state with acceptor photobleaching. (E) Long-lived stable FRET state with acceptor photobleaching. This figure is reproduced with minimal modification from Liauw et al.11. Please click here to view a larger version of this figure.
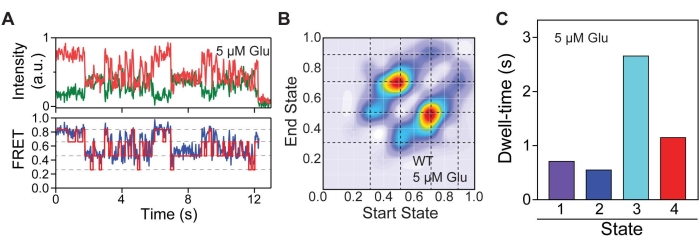
Figure 5: Conformational state identification. (A) Representative FRET trace with idealized fit overlaid (red) along with the corresponding donor and acceptor signal. (B) Transition density heatmap highlighting the most frequent conformational transitions undergone by 548UAA. Dashed lines indicate FRET states. (C) Average dwell-times of each conformational state. This figure is reproduced with a minimal modification from Liauw et al.11. Please click here to view a larger version of this figure.
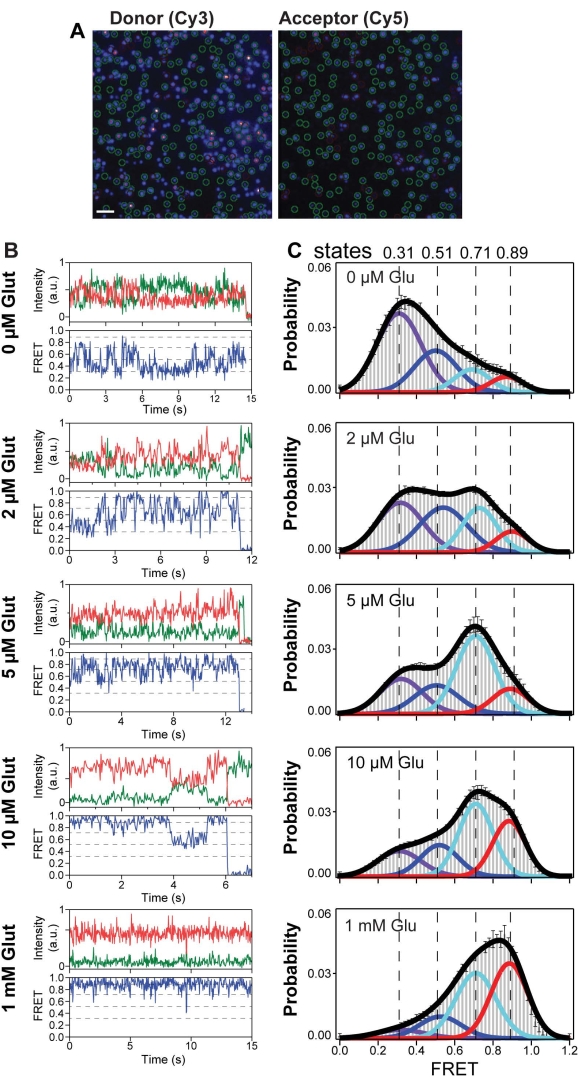
Figure 6: Single-molecule FRET reveals four conformational states of mGluR2 CRD. (A) Representative frame from a single-molecule movie with the donor channel (Cy3) on the left and the acceptor channel (Cy5) on the right. Molecules selected by analysis software for downstream processing are indicated by green circles. Scale bar = 3 µm. (B) Example single-molecule time traces of the 548UAA at different glutamate concentrations. Donor (green) and acceptor (red) intensities and the corresponding FRET (blue) are shown. Dashed lines represent four distinct FRET states. (C) smFRET population histograms in a range of glutamate concentrations. Data represent mean ± SEM. of N = 3 independent experiments. This figure is reproduced with a minimal modification from Liauw et al.11. Please click here to view a larger version of this figure.
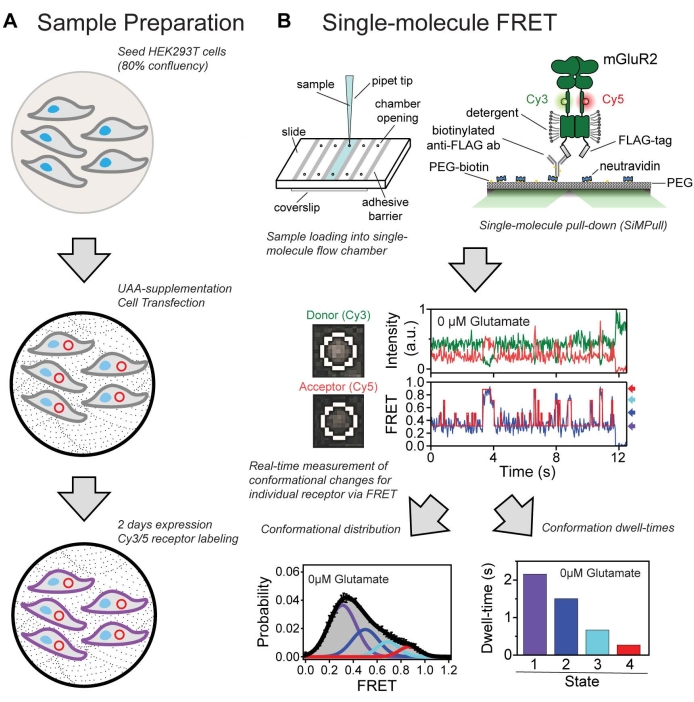
Figure 7: Summary of the overall protocol. (A) Workflow for growing and labeling cells expressing a protein containing an unnatural amino acid. (B) Single-molecule FRET experimental and analysis workflow used to identify conformational states and characterize the dynamic properties of the CRD domain in mGluR2. Please click here to view a larger version of this figure.
| Component | Volume/Reaction (μL) |
| Reduced Serum Medium | 100 |
| Transfection reagent | 4.4 |
| Component | Volume/Reaction (μL) |
| Reduced Serum Medium | 100 |
| P3000 | 4 |
| Construct/Component Name | Concentration (ng/μL) | Volume/well (12-well) (μL) | Wells (#) | DNA Added (μL) |
| tRNA/synthetase | 1000 | 1 | 1 | 1 |
| Amber codon containing protein | 1000 | 1 | 1 | 1 |
Table 1: Reagents for the transfection of the HEK 293 T cell.
| Reagents | Volume to add (µL) | Stock conc (mM) | Final Conc (mM) |
| 1x RB | 655.5 | ||
| BTTES | 10.5 | 50 | 0.75 |
| CuSO4 | 5.25 | 20 | 0.15 |
| NaAsc | 17.5 | 100 | 2.5 |
| AminoG | 8.75 | 100 | 1.25 |
| Cy3-Alkyne (10 mM) | 1.25 | 10 | 0.018 |
| Cy5-Alkyne (10 mM) | 1.25 | 10 | 0.018 |
Table 2: Composition of the labeling solution (click chemistry).
Supplementary File 1: Composition of various buffers used in this study. Please click here to download this File.
