The animal use and procedures were approved by the St. Jude Children's Research Hospital Committee on the Use and Care of Animals.
1. Preparing the solutions
- Prepare L929-conditioned media.
- Plate 1 × 106 L929 cells (see Table of Materials) in a 182 cm2 tissue culture flask containing 50 mL of L929 culture media (see Table 1 for the preparation of the media).
- Grow the cells in a humidified incubator at 37 °C with 5% CO2.
- After 7 days, collect the supernatant, and filter using a 0.45 µm filter. Prepare 50 mL aliquots (store frozen aliquots at −80 °C for up to 1 year).
- Prepare 500 mL of bone marrow-derived macrophage (BMDM) culture media (Table 1).
- Prepare 500 mL of BMDM stimulation media (Table 1).
- Prepare 500 mL of media for infection (Table 1).
- Prepare 100 mL of 1 M Tris buffer (Table 1).
- Prepare 4x sodium dodecyl sulfate (SDS) buffer (Table 1).
- Prepare 1 mL of 1 M 1,4-dithiothreitol (DTT, Table 1).
- Prepare 40 mL of caspase lysis buffer (Table 1).
- Prepare 100 mL of 1.5 M Tris buffer (Table 1).
- Prepare 100 mL of a 10% (wt/vol) SDS solution (Table 1).
- Prepare 50 mL of a 2x radioimmunoprecipitation assay (RIPA) buffer (Table 1).
- Prepare a 5 mg/mL LPS solution (Table 1).
- Prepare a 0.5 M ATP solution (Table 1).
- Prepare the western blotting solutions.
- Prepare 1 L of 5x running buffer stock (Table 1).
- Prepare 1 L of 10x transfer buffer stock (Table 1).
- Prepare 1 L of Tris-buffered saline with Tween 20 (TBST, Table 1).
- Prepare 100 mL of a 5% (wt/vol) skim milk blocking solution (Table 1).
- Prepare the primary antibody solutions.
- Prepare 10 mL of caspase-1 primary antibody (Table 1).
- Prepare 10 mL of caspase-11 primary antibody (Table 1).
- Prepare 10 mL of caspase-3 primary antibody (Table 1).
- Prepare 10 mL of caspase-7 primary antibody (Table 1).
- Prepare 10 mL of caspase-8 primary antibody (Table 1).
- Prepare 10 mL of caspase-9 primary antibody (Table 1).
- Prepare 10 mL of HRP-conjugated β-actin primary antibody (Table 1).
- Prepare the secondary antibody solutions.
- Prepare 10 mL of anti-rabbit secondary antibody (Table 1).
- Prepare 10 mL of anti-mouse secondary antibody (Table 1).
- Prepare 10 mL of anti-rat secondary antibody (Table 1).
2. Isolating bone marrow-derived macrophages
NOTE: For this protocol, 6-10-week-old wild-type mice with intact PCD pathways or mutant mice with the PCD regulators, effectors, or molecules of interest deleted or altered can be used.
- Euthanize a mouse in a CO2 chamber with a flow rate that displaces 10%-30% of the cage volume per min for 2-3 min. Then, perform a secondary euthanasia method, such as cervical dislocation. Follow all additional facility-, institution-, and government-specific guidelines and regulations wherever applicable.
- Dissect the mouse to retrieve the hind leg bones.
- Pin the mouse on its back so the abdomen is exposed. Spray with 70% (vol/vol) ethanol to sterilize the hind legs and abdomen.
CAUTION: Ethanol is flammable. Keep it away from open flames. - Use scissors to make an incision at the midline of the abdomen; continue cutting toward the legs to make the femurs visible.
- Take the right rear leg and pull the skin away from the body toward the midline. Detach the leg from the body by severing the adductor muscles toward the midline; then, cut the leg between the hip joint and the spine. Next, cut the paw off distal to the ankle, and remove excess tissue from the bone by peeling off the skin and using slightly opened scissors to strip off the calf tissue.
- Place the leg on a 70% (vol/vol) ethanol-soaked towel and dissect the tibia and the femur.
- Clasp the tibia and femur each with a separate set of forceps. Gently press the tibia against the knee joint's natural direction; this will cause the tibia to break at the knee.
- Use the forceps and dissecting scissors if needed to remove any remaining hanging tissue. Save the tibia for later use by placing it on the ethanol-soaked towel.
- Collect the femur in the same way, by snapping off the knee.
- Repeat step 3 and 4 above for the left leg to remove it from the body and dissect the tibia and femur.
- Spray the bones with 70% (vol/vol) ethanol.
- Clean off the bones by placing them on a clean 70% (vol/vol) ethanol-soaked towel, squeezing the fleshy part in the towel, and rubbing the towel against the bone to remove excess tissue.
- Pin the mouse on its back so the abdomen is exposed. Spray with 70% (vol/vol) ethanol to sterilize the hind legs and abdomen.
- Once both femurs and both tibias are clean, spray all four bones with 70% (vol/vol) ethanol. Collect the bones in a sterile Petri dish and rinse them with 10 mL of BMDM culture media by gently swishing the media in the dish.
- Fill a 10 mL syringe with 10 mL of fresh BMDM culture media and attach a 25 G needle.
- Pick up the tibia using forceps; then, cut the ankle joint at a ~45° angle.
- Flush the bone marrow from the tibia.
- Hold the tibia over a 50 mL tube, with the narrow end of the tibia pointing down. Dispense the media over the tibia from the filled syringe.
- Insert the needle (gently at first) at the top end of the marrow and dispense the media.
- Remove the needle and then insert again. Use short, high-pressure pushes to dispense the media and move the needle into the marrow.
- Once the media begins to flow out from the bottom of the bone, use short, high-pressure pushes to continue to flush the cells out. Monitor the color of the bone during this process, and when the bone is white, discard it.
- Do this for both tibias.
- Repeat steps 5 and 6 above for the femurs, cutting at the hip joint as the tibia was cut at the ankle joint. Use the same 50 mL tube to collect the BMDM culture media as the marrow is flushed.
- Once all four bones have been flushed, aspirate the marrow and media from the 50 mL tube up and down 3x through an 18 G needle on a 10 mL syringe, rinsing the sides of the tube each time to disperse the marrow.
- Adjust the final volume in the 50 mL tube to 30 mL with BMDM culture media, and ensure the cells are thoroughly suspended.
- Use a 70 µm cell strainer to filter the BMDM culture media from the 50 mL tube.
- Plate the resulting cell suspension, which contains the bone marrow progenitor cells, into three 150 mm tissue culture dishes by adding 10 mL (or ~20 × 106 cells) to each. Then, add an additional 10 mL of BMDM culture media to each dish. Incubate in a humidified incubator at 37 °C.
3. Differentiating the BMDMs and plating for the experiments
- Incubate the plated bone marrow progenitor cells at 37 °C for 3 days. Then, remove each dish, and add an additional 5-8 mL of BMDM culture media (Table 1). Return to the incubator at 37 °C.
- On day 5 after the initial plating, remove each dish, and add an additional 5 mL of BMDM culture media. Return to the incubator at 37 °C.
- On day 6, remove each dish, and discard the media. Then, add 10 mL of cold (stored at 4 °C) PBS to wash once. Discard the 10 mL of cold PBS wash. Then, add 10 mL of fresh, cold PBS to each dish, and incubate each dish on ice for 5 min.
- Using a cell scraper, gently scrape the cells from all three dishes into one 50 mL tube. Gently spin down the cells at 270 × g at 4 °C for 5 min; then, discard the supernatant.
- Add 20 mL of BMDM culture media, pipette up and down to resuspend the pellet, and count the cells.
NOTE: It is expected that each mouse will yield approximately 60 × 106-100 × 106 cells. - Plan out the 12-well plate layout for the desired in vitro cell death/inflammation stimulation assay. Plan to plate 1 × 106 cells per well. For each planned stimulation, include at least three biological replicates, and plate one set of wells to be harvested in caspase lysis buffer and a second set of wells to be harvested in RIPA buffer.
- Plate 1 × 106 cells in 1 mL of BMDM culture media per well in 12-well plates. Culture overnight in a humidified incubator at 37 °C before proceeding to the cell death/inflammation evaluation. After overnight incubation, remove the media, and add 1 mL of warm (37 °C) PBS to each well to wash the cells.
- Remove the PBS wash, and add 500 µL of BMDM stimulation media with antibiotics (if performing non-bacterial stimulations) or BMDM stimulation media without antibiotics (if performing bacterial stimulations) (Table 1). Incubate for 2 h before moving to step 4 for in vitro stimulation/infection.
4. Stimulating or infecting the cells
CAUTION: The agents included in this protocol are potentially pathogenic and should be handled with the appropriate precautions in a biosafety level 2 (BSL2) facility with approval from the relevant institutional and governmental authorities.
- Stimulate the BMDMs to activate cell death with the trigger of interest.
NOTE: For the purposes of this protocol, influenza A virus (IAV), herpes simplex virus 1 (HSV1), Francisella novicida, and LPS + ATP are used for illustration, but other triggers can be used.- Example stimulation 1: Infect with IAV (A/Puerto Rico/8/34, H1N1 [PR8]) (constructed as per Hoffmann et al.75; the viral titer for the multiplicity of infection [MOI] determination is calculated by a plaque assay in MDCK cells):
- Calculate the volume of virus needed for infection at a multiplicity of infection (MOI) of 20 plaque-forming units (PFU) using equation (1) and equation (2):

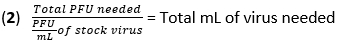

- Remove the media from the BMDMs and wash the cells once with 500 µL of PBS. Add 450 µL of IAV (20 MOI) in high-glucose DMEM without heat-inactivated (HI)-FBS to each well. Incubate the plates at 37 °C for 1 h in a humidified incubator to allow absorption.
- Remove the plates and add 50 µL of HI-FBS. Return the plates to the incubator at 37 °C. Incubate for a total of 12 h.
- Calculate the volume of virus needed for infection at a multiplicity of infection (MOI) of 20 plaque-forming units (PFU) using equation (1) and equation (2):
- Example stimulation 2: Infect with HSV1 (HF strain; cultured as described previously44; the viral titer for MOI determination is calculated by a plaque assay in Vero cells):
- Calculate the volume of virus needed for infection at an MOI of 10 PFU using equation (1) and equation (2) from step 1 in the IAV infection section above.

- Remove the media from the BMDMs. Add 450 µL of HSV1 (MOI 10) in high-glucose DMEM without HI-FBS to each well. Incubate the plates at 37 °C for 1 h in a humidified incubator to allow absorption.
- Remove the plates, and add 50 µL of HI-FBS. Return the plates to the incubator at 37 °C. Incubate for a total of 12 h.
- Calculate the volume of virus needed for infection at an MOI of 10 PFU using equation (1) and equation (2) from step 1 in the IAV infection section above.
- Example stimulation 3: Infect with F. novicida (U112 strain; cultured as described previously44 under aerobic conditions at 37 °C in BBL Trypticase Soy Broth supplemented with 0.2% L-cysteine overnight. Then, subculture the bacteria at a ratio of 1:10 at 37 °C for another 4 h in fresh medium before taking the optical density (OD) at 600 nm using the fresh medium as a blank. An OD value of 1 equates to 1 × 109 colony-forming units (CFU) per mL.)
- Calculate the volume of F. novicida needed for infection at an MOI of 50 CFU using equation (3) and equation (4):

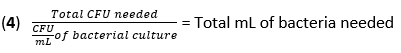
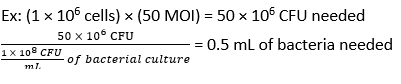
- Remove the media from the BMDMs. Add 500 µL of F. novicida (MOI 50) in BMDM stimulation media without antibiotics to each well. Incubate the plates at 37 °C for 4 h in a humidified incubator to allow absorption.
- Wash the cells three times with warmed (37 °C) PBS and add 500 µL of BMDM stimulation media containing 50 µg/mL gentamycin. Return the plates to the incubator at 37 °C. Incubate overnight (16 h).
- Calculate the volume of F. novicida needed for infection at an MOI of 50 CFU using equation (3) and equation (4):
- Example stimulation 4: Stimulate with LPS + ATP.
- Remove the media from the BMDMs. Add 500 µL of BMDM stimulation media with antibiotics (Table 1) containing 100 ng/mL LPS to each well. Incubate the plates at 37 °C for 3.5 h in a humidified incubator.
- Add 5 µL of 0.5 M ATP stock solution (Table 1) to each well. Return the plates to the incubator at 37 °C. Incubate for 30 min.
- Example stimulation 1: Infect with IAV (A/Puerto Rico/8/34, H1N1 [PR8]) (constructed as per Hoffmann et al.75; the viral titer for the multiplicity of infection [MOI] determination is calculated by a plaque assay in MDCK cells):
5. Collecting the combined supernatant and protein lysate to be used for caspase western blots
- After 4 h, 12 h, or 16 h of incubation (the specific timing is dependent on the trigger used), remove the plate from the incubator.
- Remove 150 µL of the supernatant; discard or save this for other supernatant analyses (e.g., enzyme-linked immunosorbent assay [ELISA]). Do not remove the remaining supernatant.
- Create the protein collection solution by combining 50 µL of caspase lysis buffer + 100 µL of 4x SDS buffer per well (Table 1). Then, add 150 µL of the mix to each well.
- For each well, pipette the mixture up and down to collect the lysed cells and supernatant. While pipetting, also scrape the bottom of the well with the pipette tip to disrupt the cells. After scraping and pipetting, collect the protein lysate into labeled 1.5 mL tubes.
- Use a heat block to heat all the tubes to 100 °C for 12 min.
- Pellet any insoluble components by centrifuging at 14,500 × g for 30 s at room temperature.
NOTE: This is a pause point – the protein from the combined supernatant and protein lysates can either be used immediately or stored at −20 °C for up to 2 months or at −80 °C for up to 6 months until ready to use.
6. Collecting the protein lysate to be used for caspase western blots
- After 4 h, 12 h, or 16 h of incubation (the specific timing is dependent on the trigger used), remove the plate from the incubator. Remove all the supernatant; discard or save this for other supernatant analyses (e.g., ELISA).
- Create the 1x RIPA buffer by diluting the 2x RIPA stock solution (Table 1) in an equal volume of deionized water. Then, add one phosphatase inhibitor tablet and one protease inhibitor tablet, and allow them to dissolve. Add 150 µL of 1x RIPA buffer and 50 µL of 4x SDS to each well.
- For each well, pipette the mixture up and down to collect the lysed cells. While pipetting, also scrape the bottom of the well with the pipette tip to disrupt the cells. After scraping and pipetting, collect the protein lysate into labeled 1.5 mL tubes.
- Use a heat block to heat all the tubes to 100 °C for 12 min.
- Pellet any insoluble components by centrifuging at 14,500 × g for 30 s at room temperature.
NOTE: This is a pause point – the protein lysates can either be used immediately or stored at −20 °C or −80 °C until ready to use.
7. Performing western blotting using the lysates collected from the BMDMs following the steps above or from tissue homogenates
NOTE: If using tissue, it can be homogenized by hand or through a power-driven tissue homogenizer. The protocol by Simpson76 provides a detailed description of tissue homogenization.
- Prepare 1x running buffer: Combine 200 mL of the 5x running buffer stock (Table 1) and 800 mL of deionized water. Make this 1x running buffer just before each experiment.
- Prepare the electrophoresis apparatus with a 12% (wt/vol) polyacrylamide gel with 10 wells. Fill the electrophoresis apparatus with 1x running buffer. Then, remove the gel comb.
NOTE: To analyze caspase-1, caspase-11, caspase-3, caspase-7, caspase-8, and caspase-9 for each sample, six gels will be needed. - For caspase-1, caspase-3, caspase-7, and caspase-8 blots, plan to use 30 µL of the combined supernatant and protein lysate in caspase lysis buffer or the tissue homogenate. For caspase-11 and caspase-9 blots, plan to use 20 µL of the protein lysate in RIPA buffer or the tissue homogenate. If the samples have been stored at −20 °C or −80 °C, thaw them on ice first.
- For all the samples, heat to 100 °C for 5 min, and centrifuge at 14,500 × g for 30 s at 4 °C before loading. Then, slowly load 20 or 30 µL of the sample into each lane. Avoid having any sample overflow into the other lanes. To evaluate all six caspases at once, use the same procedure to load the appropriate samples into each of the six gels.
- Connect the electrophoresis apparatus to the power source. Then, set the power to 80 V for 20 min to begin the gel run, and then adjust the power to 130 V for 45-60 min.
- Watch the dye front carefully, and turn the power off when the dye front is at the bottom of the gel but has not yet been pushed out of the gel.
- While the gel is running, prepare 1x transfer buffer by combining 700 mL of deionized water, 100 mL of the 10x transfer buffer stock (Table 1), and 200 mL of methanol. Make the 1x solution fresh each time.
NOTE: Use caution as methanol is flammable. Perform the transfer buffer preparation away from open flames. - Remove the gel from the electrophoresis apparatus gently by using the gel releaser.
- Set up one transfer stack for each gel.
- Activate a PVDF membrane by soaking it in methanol for 1 min.
- Pre-wet two pieces of filter paper, the gel, and the PVDF membrane in transfer buffer for 5 min. Keep the PVDF membrane and gel in separate containers during this 5 min incubation.
- Assemble the transfer stack on the semi-dry system. Beginning on the bottom platinum anode side, place one piece of filter paper, the PVDF membrane, the gel, and finally one piece of filter paper. Gently roll out or press out air bubbles between the layers, and close the top of the system. Ensure that the safety cover is secured before proceeding further.
- Connect to the power source. Set the power to 25 V for 45 min.
- After the transfer is complete, disassemble the transfer stack, and collect the membrane; place it in a square Petri dish (incubation tray).
- Perform membrane blocking by adding 15 mL of a 5% (wt/vol) skim milk solution (Table 1). Incubate the membrane on a rocking shaker at 50 rpm to 70 rpm at room temperature for 1 h.
NOTE: This is a pause point – the membrane can either be removed after 1 h or stored in blocking solution at 4 °C overnight. - After 1 h or overnight incubation, remove the blocking solution. Add 10 mL of the diluted antibody solution (anti-caspase-1 antibody, anti-caspase-11 antibody, combined anti-caspase-3 and anti-cleaved caspase-3 antibody, combined anti-caspase-7 and anti-cleaved caspase-7 antibody, combined anti-caspase-8 and anti-cleaved caspase-8 antibody, or anti-caspase-9 antibody) (Table 1). Place on a rocking shaker at 50 rpm to 70 rpm to incubate at room temperature for 2 h or at 4 °C overnight (16 h).
- Collect the antibody solution (reuse up to 3x or discard it), and wash by adding 15 mL of TBST (Table 1) to the membrane on a rocking shaker at 50 rpm to 70 rpm at room temperature for 10 min. Discard the TBST.
- Repeat the wash with 15 mL of TBST following step 14 a total of 3x.
- Add 10 mL of the diluted secondary HRP-conjugated antibody solution (anti-rabbit for blots stained with primary antibodies against caspase-3, caspase-7, caspase-8, or caspase-9; anti-mouse for blots stained with primary antibody against caspase-1; anti-rat for blots stained with primary antibody against caspase-11) (Table 1). Incubate on a rocking shaker at 50 rpm to 70 rpm at room temperature for 1 h.
- Remove the antibody solution, and wash by adding 15 mL of TBST to the membrane on a rocking shaker at 50 rpm to 70 rpm at room temperature for 10 min. Discard the TBST.
- Repeat the wash with 15 mL of TBST following step 17 a total of 3x.
- Add 10 mL of the high-sensitivity HRP substrate to the membrane. Let it sit at room temperature in the dark for 1 min.
- Remove the membrane from the substrate. Proceed directly to the imaging, using a chemiluminescence imager with the accessory white trans tray inserted in the lower position. Expose the membrane using the auto exposure mode (generally ~1-2 min of exposure time).
- Using the membrane from the caspase-9 or caspase-11 blotting (i.e., a membrane with RIPA lysate samples), add 10 mL of stripping buffer, and incubate on a rocking shaker at 50 rpm to 70 rpm at room temperature for 5 min.
- Discard the stripping buffer, and wash by adding 15 mL of TBST to the membrane on a rocking shaker at 50 rpm to 70 rpm at room temperature for 10 min. Discard the TBST.
- Repeat the wash with 15 mL of TBST following step 22 a total of 3x.
- Perform membrane blocking by adding 15 mL of a 5% (wt/vol) skim milk solution. Incubate the membrane on a rocking shaker at 50 rpm to 70 rpm at room temperature for 1 h.
NOTE: This is a pause point – the membrane can either be removed after 1 h or stored in blocking solution at 4 °C overnight. - After 1 h or overnight incubation, remove the blocking solution. Add 10 mL of the diluted anti-β-actin (HRP-conjugated) antibody solution. Place on a rocking shaker at 50 rpm to 70 rpm to incubate at room temperature for 1.5 h.
- Remove the antibody solution and wash by adding 15 mL of TBST to the membrane on a rocking shaker at 50 rpm to 70 rpm at room temperature for 10 min. Discard the TBST.
- Repeat the wash with 15 mL of TBST following step 26 a total of 3x.
- Add 10 mL of the standard-sensitivity HRP substrate to the membrane. Let it sit at room temperature in the dark for 1 min.
- Proceed directly to the imaging, using a chemiluminescence imager with the accessory white trans tray inserted in the lower position. Expose the membrane using the auto exposure mode (generally <1 min of exposure time).
PANoptosis has been observed in response to numerous bacterial, viral, and fungal infections and other inflammatory stimuli, as well as in cancer cells44,48,49,50,51,52,53,54,56,57,58,60,61,62. In these cases, the activation of multiple caspases has been reported. Using the example stimulations in this protocol that are known to induce PANoptosis, namely IAV, HSV1, and F. novicida44,48,50,51,53,57, it is expected that cleavage may be observed as a surrogate for the activation of multiple caspases (Figure 2A-C). Additionally, LPS + ATP is included as a control; this combination is a canonical NLRP3 inflammasome trigger that is expected to induce caspase-1 activation. The activation of caspase-1 can be visualized in response to each of these triggers by the cleavage of the p45 pro-form to the p20 active form (Figure 2D). The p20 form can then cleave GSDMD and initiate cell death19.
Similarly, the activation of the apoptotic initiator caspase, caspase-8, is observed, as denoted by the presence of the p18 cleaved form (Figure 2A–D). The weak activation of caspase-9 can also be observed in some cases with these stimuli (Figure 2A–D). Downstream of these initiators, the effector caspases caspase-3 and caspase-7 are activated; this activation is observed as the formation of the p17/19 cleaved form of caspase-3 and the p20 cleaved form of caspase-7 (Figure 2A–D). To date, the activation of caspase-11 has not been widely evaluated in PANoptosis, but its activation has been observed in some contexts54. Caspase-11 remains an important inflammatory caspase, and its activation can be observed by the formation of its p26 cleaved form. While some low-level caspase-11 activation is seen, robust activation is not observed in response to IAV, HSV1, or F. novicida infection or LPS + ATP stimulation (Figure 2A–D).
Cells lacking upstream PANoptosis sensors do not undergo cell death in response to PANoptosis-inducing stimuli. For example, IAV infection induces the formation of the ZBP1-PANoptosome48,51,53,57, and Zbp1-/- cells are protected from cell death during IAV infection (Figure 3A). Similarly, HSV1 and F. novicida infections induce the formation of the AIM2-PANoptosome44, and Aim2-/- cells fail to undergo robust cell death in response to these infections (Figure 3B,C). In cells that are deficient in the upstream sensor necessary for PANoptosome formation, the activation of caspases is significantly reduced or even eliminated, as can be seen by the reduction in the intensity of the cleaved bands in the western blots (Figure 2A–C). Similarly, cells lacking upstream inflammasome sensors are protected from cell death in response to their respective stimuli, and Nlrp3-/- cells do not undergo cell death in response to LPS + ATP (Figure 3D). These cells also show reduced caspase activation (Figure 2D).
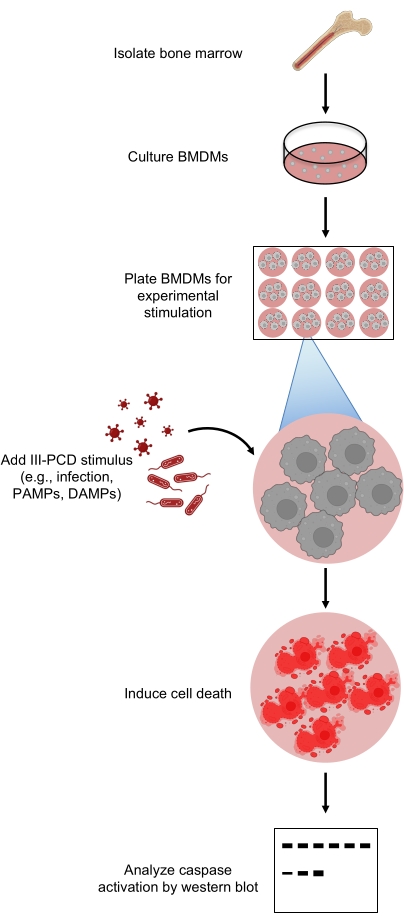
Figure 1: Overview of the experimental procedure. Bone marrow is isolated and differentiated into bone marrow-derived macrophages. These cells are then stimulated with a trigger that activates innate immune signaling and cell death, such as infection or an inflammatory stimulus. After the cells activate and begin to undergo PCD, the lysate is collected and analyzed by western blotting to monitor caspase cleavage to their active forms. Abbreviations: BMDMs = bone marrow-derived macrophages; DAMPs = damage-associated molecular patterns; III-PCD = inflammatory innate immune programmed cell death; PAMPs = pathogen-associated molecular patterns; PCD = programmed cell death. Please click here to view a larger version of this figure.
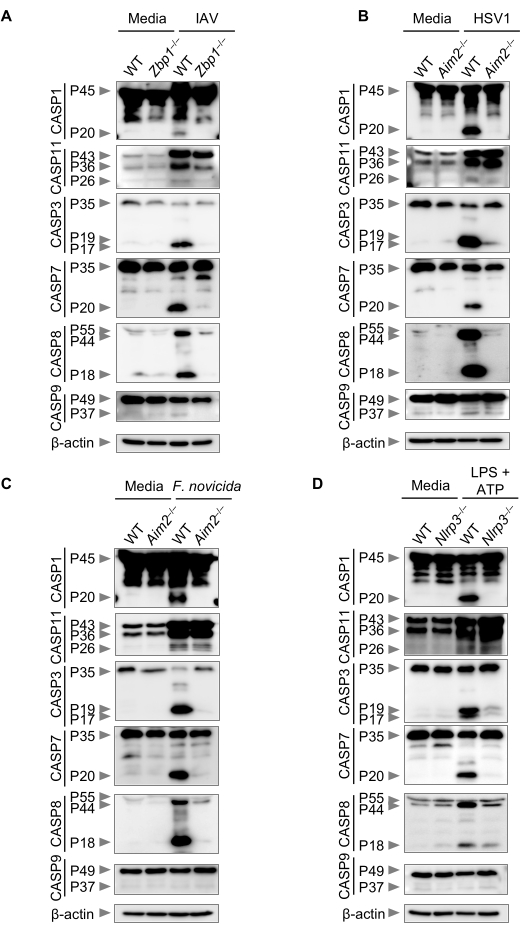
Figure 2: Activation of caspases in response to stimuli. Caspase activation from cell culture lysates can be assessed by evaluating the size of the products run on an electrophoretic gel. (A–D) Immunoblot analysis of pro- (P45) and activated (P20) CASP1, pro- (P43) and cleaved (P36 and P26) CASP11, pro- (P35) and cleaved (P17/P19) CASP3, pro- (P35) and cleaved (P20) CASP7, pro- (P55) and cleaved (P44 and P18) CASP8, and pro- (P49) and cleaved (P37) CASP9 in WT and Zbp1−/−, WT and Aim2−/−, or WT and Nlrp3−/− BMDMs after (A) IAV infection, (B) HSV1 infection, (C) Francisella novicida infection, or (D) LPS + ATP stimulation. Images are representative of three independent experiments. Abbreviations: BMDMs = bone marrow-derived macrophages; CASP1 = caspase-1; CASP3 = caspase-3; CASP7 = caspase-7; CASP8 = caspase-8; CASP9 = caspase-9; CASP11 = caspase-11; HSV1 = herpes simplex virus 1; IAV = influenza A virus; LPS = lipopolysaccharide; WT = wild-type. Please click here to view a larger version of this figure.
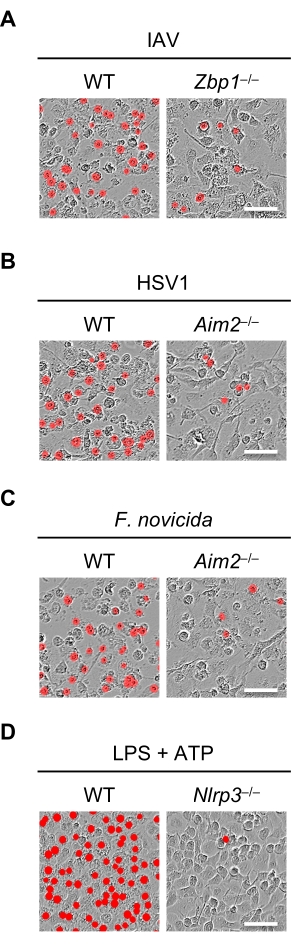
Figure 3: Inhibition of cell death when caspase activation does not occur. (A–D) Representative images of cell death in WT and Zbp1−/−, WT and Aim2−/−, or WT and Nlrp3−/− BMDMs after (A) IAV infection, (B) HSV1 infection, (C) Francisella novicida infection, or (D) LPS + ATP stimulation. The red mask denotes dead cells. Scale bars = 50 µm. Images are representative of three independent experiments. Abbreviations: BMDMs = bone marrow-derived macrophages; HSV1 = herpes simplex virus 1; IAV = influenza A virus; LPS = lipopolysaccharide; WT = wild-type. Please click here to view a larger version of this figure.
