1. In vitro kinetics experiment
- Preparation of DNA templates by PCR
- Set up PCR reaction(s): To prepare PCR reactions, combine the following reagents in a thin-walled PCR tube:
33 µL of double-distilled water (ddH2O)
10 µL of 5x buffer for high-fidelity DNA polymerase
5 µL of 2 mM each deoxyribonucleoside triphosphate (dNTP)
0.5 µL of 40 µM forward primer
0.5 µL of 40 µM reverse primer
0.5 µL (10-100 ng) of DNA template (for Spinach2 PCR only; Broccoli primers overlap)
0.5 µL of high-fidelity DNA polymerase (add last)
NOTE: Synthetic oligonucleotides are often shipped dry. To prepare stock solutions, add a known volume (100 µL recommended) of ddH2O, and measure the A260 of that solution to determine the concentration by Beer's law, where the extinction coefficient can be calculated by nearest-neighbor rules online. This stock solution can then be used to make dilutions appropriate for use in PCR. - Run the thermocycler protocol.
- Use the following thermocycler protocol to amplify full-length Spinach2 and Broccoli DNA templates:
Initial denaturation:98 °C for 2 min
35 cycles:
Denaturation:98 °C denaturation for 15 s
Annealing:72 °C for 30 s
Extension:72 °C for 30 s
Final extension: 72 °C for 5 min. - After the reaction, analyze a small aliquot of the product by 2% agarose gel along with a low molecular weight ladder (size range: 25-766 nucleotides) to confirm the presence of the desired DNA product.
- Purify the product by commercially available gel extraction or PCR clean-up kits and elute with ddH2O or the buffer provided by the manufacturer.
NOTE: Be sure when choosing a PCR clean-up kit that the column molecular weight cutoff is low enough to retain T7-Broccoli (81 nucleotides), or else the PCR product will be lost.
NOTE: Optional pause point: store the DNA at −20 °C.
- Use the following thermocycler protocol to amplify full-length Spinach2 and Broccoli DNA templates:
- Set up PCR reaction(s): To prepare PCR reactions, combine the following reagents in a thin-walled PCR tube:
- Preparation of Spinach2 and Broccoli RNA by in vitro transcription (IVT)
- Set up the transcription reaction(s).
- To prepare a 100 µL transcription reaction, combine the following reagents in a 1.5 mL microcentrifuge tube: 10 µL of 10x transcription buffer + 20 µL of 10 mM ribonucleoside triphosphates (rNTPs) + 1-64 µL of DNA template (total of 1 µg) + 2 µL of inorganic pyrophosphatase + ddH2O to 98 µL + 2 µL of T7 RNA polymerase (add last).
- Incubate this reaction for 4 h at 37 °C. Quench the reaction by adding 100 µL of 2x urea gel loading buffer (2x ULB), composed of 20% sucrose, 0.1% sodium dodecyl sulfate (SDS), 1x Tris-Borate-EDTA (1x TBE) buffer, and ~18 M urea.
NOTE: Optional pause point: store the quenched reaction at −20 °C.
- Set up the transcription reaction(s).
- Polyacrylamide gel electrophoresis (PAGE) purification of RNA
- PAGE purification of Spinach2 and Broccoli RNA
CAUTION: Non-polymerized (liquid or powdered) acrylamide is extremely toxic. If weighing out powdered acrylamide, do so in a fume hood. Always wear proper protective equipment and immediately remove gloves contaminated with acrylamide powder or solution, washing hands thoroughly. If acrylamide comes in direct contact with the skin, wash the exposed area for at least 15 min with soap and water. If acrylamide comes in direct contact with the eyes, flush them with water for 15 min.- Prepare the PAGE gel: To remove unwanted abortive transcripts and unreacted rNTPs from the full-length product, prepare a 6% urea-polyacrylamide gel. In general, 28 cm x 16.5 cm x 1.5 mm gels can be used with an 8-well comb. Set up the gel and electrophoresis equipment, using 1x TBE buffer to fill the reservoirs.
- Load RNA sample(s) in the PAGE gel: Load the gel with one quenched 200 µL reaction per lane. In a separate lane, load 2x ULB with tracker dyes xylene cyanol and bromophenol blue, which migrate in the gel at 106 nucleotides and 26 nucleotides, respectively15. Leave an empty lane between each sample to avoid potential contamination in the next steps.
- Run the PAGE gel: To separate 95-nt Spinach2 and 49-nt Broccoli from their respective truncated products, run the gel for 1.5-2 h at 25 W, at which point the bromophenol blue dye will have migrated ~5/6 of the gel length.
- Visualize the RNA sample(s) in the PAGE gel: Disassemble the glass plates around the gel and cover the gel in plastic wrap on both sides, labeling the lanes on the wrap. Visualize RNA bands in a dark room by UV shadowing by placing the wrapped gel on a fluorescent TLC plate under UV light. Quickly outline the edge of the RNA bands corresponding to the product with a marker and switch off the UV lamp to minimize damage from UV exposure.
- Excise and extract the RNA sample(s) from the PAGE gel: With a fresh razor blade for each sample, excise the desired product bands, dice into ~1 mm cubes, and add the gel pieces to a 2 mL microcentrifuge tube with 500 µL of crush soak buffer to extract RNA on a rotator for either 2 h at room temperature (RT) or overnight at 4 °C.
NOTE: Optional pause point: the sample may be stored at −20 °C. - Precipitate the RNA
- To separate the gel pieces from the extracted RNA in buffer, centrifuge the sample at 13,000 x g for 20 min at 4 °C, and then use a narrow-tip pipette to extract the supernatant and load it into a new 2 mL microcentrifuge tube.
- To precipitate the RNA, add 1.5 mL of ice-cold ethanol and 1 µL of 20 mg/mL glycogen, vortex, and store for at least 1 h at −20 °C or −80 °C.
NOTE: Optional pause point: the RNA may be stored at −20 °C for a few months.
- Collection of RNA precipitate: Pellet the precipitated RNA by centrifugation at 13,000 x g for 20 min at 4 °C. Remove the supernatant and allow the remaining ethanol to evaporate under open air (~1 h) before resuspending the pellet in 30 µL of ddH2O or 1x TE buffer.
NOTE: This process typically results in a final RNA concentration of ~10 µM.
NOTE: Optional pause point: the RNA sample may be stored at −20 °C for a few months.
- Determine the RNA stock concentrations.
- Prepare an RNA aliquot for the hydrolysis reaction.
- To perform this assay, first, use a UV/Vis nano spectrophotometer to determine the A260 of the stock RNA sample and make a diluted aliquot of the sample to ~10 absorbance units (AU) in ddH2O.
- Prepare the following reaction in a 0.5 mL PCR tube: 16 µL of ddH2O + 2 µL of 10x Na2CO3 buffer + 2 µL of RNA aliquot diluted to ~10 AU. Incubate the reaction for 90 min at 95 °C, and then allow it to cool to RT.
- Determine the RNA concentration using nucleotide absorbances: Measure the A260 of this sample with a UV/Vis nano spectrophotometer and calculate the RNA concentration using the following formula:
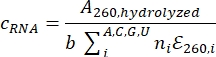
where c is the concentration of RNA, b is the path length, i is the specific nucleotide (A, C, G, or U), ni is the frequency of nucleotide i in the RNA sequence, and εi is the molar extinction coefficient of nucleotide i. To determine the original stock RNA concentration, multiply c by the dilution factor.
NOTE: Optional pause point: The RNA may be stored at −20 °C for a few months.
- Prepare an RNA aliquot for the hydrolysis reaction.
- PAGE purification of Spinach2 and Broccoli RNA
- Performing an in vitro plate reader kinetics assay
- Set up the RNA renaturation program: Create the following thermocycler protocol by selecting Create New Program > Add New Phase > Add New Step multiple times to add each of the following steps before pressing Save:
70 °C for 3 min
65 °C for 45 s
60 °C for 45 s
55 °C for 45 s
50 °C for 45 s
45 °C for 45 s
40 °C for 45 s
35 °C for 45 s
30 °C for 45 s - Renature the RNA: To renature Spinach2 and Broccoli RNAs, prepare 2 µM stocks of each RNA in ddH2O in a 0.5 mL thin-walled PCR tube, and then add an equal volume of 2x renaturation buffer (80 mM HEPES, pH 7.5 [KOH], 250 mM KCl, 6 mM MgCl2) to make 1 µM RNA solutions. Add the tubes to the thermocycler, open the saved renaturation program, and press Run.
NOTE: If a thermocycler is unavailable, the RNAs may instead be incubated on a 70 °C heat block for 3 min and then allowed to cool slowly to RT on the bench. - Prepare the binding reaction buffer: To prepare the buffer for one aptamer-dye binding reaction, make a master mix containing buffer components (69.5 µL of ddH2O + 4 µL of 1 M HEPES, pH 7.5 [KOH] + 6.2 µL of 2 M KCl + 0.3 µL of 1 M MgCl2). Depending on how many samples and replicates are needed, multiply these values by the number of samples plus one. Generally, three replicates per RNA sample is satisfactory.
- Prepare the plate reader.
- Set up the plate reader injector program: On the fluorescence plate reader, select Temp and set the desired temperature to 37 °C, making sure the temperature has equilibrated to this value well before starting kinetics experiments. Open the plate reader software, select Settings > Acquisition View, and input the following program for kinetics measurements:
Loop: For each well
Baseline setting: 60 baseline reads
SmartInject settings: 10 µL injection (which will happen after the baseline reads)
Fluorescence (or FL) reads:
Excitation: 448 nm (bandwidth: 9 nm)
Emission: 506 nm (bandwidth: 15 nm)
Cartridge: MONO (s/n 3297)
Timing:
Total read time: 10 min
Read interval: 0.5 s
PMT and Optics: 6 flashes per read
Loop: Next well - Prepare the injector
- Wash and aspirate the injector: To prepare the plate reader injector, first select Inject on the plate reader, supply a waste collection plate to the plate reader when directed, and then select Wash, and clean the injection tube following instructions on the plate reader with 1 mL volumes of ddH2O, 75% ethanol in ddH2O, and then ddH2O. Next, select Aspirate, allowing the injector to eject excess liquid.
- Prime the injector: After exiting to the previous screen, select Prime to prime the injector with two 260 µL volumes of ligand to be injected to ensure pure, concentrated ligand is added to samples during experiments-in this case, prime with 100 µM DFHBI in ddH2O.
- Set up the plate reader injector program: On the fluorescence plate reader, select Temp and set the desired temperature to 37 °C, making sure the temperature has equilibrated to this value well before starting kinetics experiments. Open the plate reader software, select Settings > Acquisition View, and input the following program for kinetics measurements:
- Perform in vitro kinetics experiments: To perform one kinetics experiment, first add 80 µL of previously prepared binding buffer master mix to one well of a 96-well clear-bottom plate, followed by 10 µL of renatured RNA. Allow this plate and the DFHBI solution in the injector to equilibrate to 37 °C for 15 min in the plate reader.
- In the plate reader software Settings under Read Area, select the well to be analyzed, and then, under the Home tab, select Run to execute the kinetics program described previously. Repeat this process until all the experiments are complete.
NOTE: Critically, kinetics experiments should be performed one well at a time to ensure the RNA kinetics are measured under identical conditions between replicates and samples. - Wash the injector: To remove the remaining DFHBI solution from the injection tube, wash the injection tube as described in step 1.4.4.2.1 with 1 mL volumes of ddH2O, 75% ethanol in ddH2O, and then ddH2O.
NOTE: Optional pause point.
- Set up the RNA renaturation program: Create the following thermocycler protocol by selecting Create New Program > Add New Phase > Add New Step multiple times to add each of the following steps before pressing Save:
- Analysis of the in vitro kinetics of fluorogenic RNA aptamers.
- Input data into the analysis software: Export experimental data as a spreadsheet to easily copy over the data for processing. In the analysis software, create a new data table in XY format. In the X column, enter each reading timepoint, with t = 0 being the time of DFHBI injection. In the Y column, enter the average fluorescence values between replicates at that respective timepoint starting at t = 0.
- Normalize and graph the data: To normalize the fluorescence values, click on Analyze > Data Processing > Normalize, and then click on OK. Choose to use the smallest and largest values of the dataset for normalization, to present results as fractions, and to Graph the Results, and then click on OK. To create a graph of the normalized averaged fluorescence over time, click the Normalize of [Dataset Name] icon and select Graph Family: XY with Points Only.
NOTE: Error bars can be useful to see in cases where a small number of time points are graphed. If these are desired, when choosing an XY format data table, select the option under "Y" to enter replicate values in side-by-side columns. Graphing the resulting table with the default options selected will produce a graph with error bars. - Perform curve fitting to obtain kinetic parameters: To fit a curve to the kinetics data, click on Analyze > Analyze Data and select Nonlinear Regression (Curve Fit) under the XY Analyses tab. Under the Model tab, click on Exponential > Two Phase Association to fit the kinetics data with the following two-phase association equation:
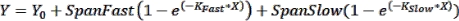
where Y is the fluorescence at time X, Y0 is the fluorescence at t = 0, KFast and KSlow are fast and slow rate constants, respectively, and SpanFast and SpanSlow are the ranges of fluorescence turn-on accounted for by the fast and slow rates, respectively (see representative results, Figure 1). Click on the Nonlin Fit tab to view rate constants, t1/2 values, and PercentFast values.
NOTE: To obtain a standard deviation for all these values, individual plate reader experiment replicates can be processed in the same way as described above.
2. Cellular kinetics experiment
- Preparation of E. coli strains
- Transform BL21 Star (DE3) E. coli cells with ~100 ng of pET31b tRNA-Spinach2 construct following the manufacturer's protocol.
NOTE: Plasmid construct is commercially available (Plasmid #79783). - Plate the cells on LB agar containing carbenicillin (Carb: 50 mg/mL) plates and incubate at 37 °C for 12-16 h. Cells containing plasmids will grow as colonies on the plate.
NOTE: Optional pause point: Transformed BL21 Star cells on plates can be stored at 4 °C wrapped in parafilm for 1 week.
- Transform BL21 Star (DE3) E. coli cells with ~100 ng of pET31b tRNA-Spinach2 construct following the manufacturer's protocol.
- Growing cells and inducing the expression of fluorogenic RNA aptamer
- Inoculate a 2 mL culture of noninducing media (NI) containing carbenicillin (Carb: 50 mg/mL) with a single colony of the transformed BL21 Star cells. Repeat this for at least three biological replicates. Incubate the cultures at 37 °C in an incubator/shaker at 250 rpm for 22-24 h.
NOTE: Optional pause point: Cells grown in NI media retain the plasmid and can be stored at 4 °C for 1 week. - After growth in NI media, dilute the culture 100x into a fresh 3 mL of ZYP-5052 autoinduction media (AI) containing carbenicillin (Carb: 50 mg/mL). Grow the cells at 37 °C in an incubator/shaker at 250 rpm for 16-18 h to induce expression.
NOTE: The typical OD600 for cultures will range from 2.0-3.3 after 18 h of growth. The optimum cell density range is between 2.5-3.0.
- Inoculate a 2 mL culture of noninducing media (NI) containing carbenicillin (Carb: 50 mg/mL) with a single colony of the transformed BL21 Star cells. Repeat this for at least three biological replicates. Incubate the cultures at 37 °C in an incubator/shaker at 250 rpm for 22-24 h.
- Performing the cellular kinetics experiment
- Set up the flow cytometer.
- Turn on the flow cytometer and computer connected to the instrument. Once logged into the software for the flow cytometer, under the Instrument tab, click on the Startup icon. Follow the steps indicated on the software screen to ensure proper instrumentation initialization.
NOTE: Some flow cytometers call the instrument startup sequence Priming. Make sure to follow the manufacturer's protocol for the flow cytometer that will be in use for the experiment. - Run a performance test (if applicable). Under the Main Menu tab, click on Performance Test. In a culture tube, add three drops of the manufacturer's performance tracking beads into 3 mL of focusing fluid.
- Place the culture tube into the sample tube lifter and raise the lifter. Prior to clicking Run Performance Test, make sure the Lot # of the tube of the tracking beads is the same as what is indicated on the Performance Test Setup screen. Click Performance Test to run the test.
- Set up the flow cytometer software for this experiment with the following acquisition parameters for single-cell fluorescence:
Excitation Laser: 488 nm
Emission Channel: GFP (also called FITC)
Acquisition Volume: 40 µL (with a total draw volume of 90 µL)
Flow Rate: 200 µL/ min
Cell Counts for each measurement: 30,000
Instrument Settings:
Voltage:
FSC: 480 V
SSC: 400 V
BL1: 540 V
BL2: 392 V
BL3: 422 V
- Turn on the flow cytometer and computer connected to the instrument. Once logged into the software for the flow cytometer, under the Instrument tab, click on the Startup icon. Follow the steps indicated on the software screen to ensure proper instrumentation initialization.
- Set up the experimental files.
- Create a new experiment file within the Experiment Explorer tab by right-clicking the flow cytometer username. Select New Experiment in the drop-down window. When a new window on the computer screen pops up, select OK.
- In the new experiment file, right-click the "Group" folder and select Add New Sample Tube. Label the sample tubes for each specific time point and replicate by right-clicking on Sample and selecting Rename in the drop-down menu. Repeat this step for the total number of replicates and time points for the intended time course of the study.
- Prepare a dilute cell solution: In a new culture tube, add 1.5 mL of 1x PBS solution. Next, add 3 µL of induced cells in AI media into the 1x PBS solution to make a dilute cell solution. Repeat this step for each biological replicate in different culture tubes.
- Measure the background fluorescence of the cells: Before adding dye, take readings of each biological replicate culture tube containing cells in 1x PBS solution. This is so that the fluorescent background of the cells is measured to observe the fold turn on over time once the dye is added.
- To take a reading, place the culture tube into the sample tube lifter, and raise the lifter by hand to the sample injection needle. Select the proper sample file within the Collection Panel tab, and click Record.
- When the run is complete, lower the sample tube lifter with the culture tube by hand. This will initiate a Rinse step that will flush the fluidic system and minimize carryover between each biological replicate sample. The data will automatically be saved to the computer after the run is completed.
- Repeat the steps within 2.3.4 to measure the cellular fluorescent background for at least three biological replicates. To move to the next sample file, select the next sample file by clicking the right arrow icon near the sample tube name underneath the Record icon.
- Measure fluorescence at time points for cells with dye.
- Add 1.4 µL of a concentrated dye stock (50 mM DFHBI-1T in DMSO) into 1x PBS solution with cells to give a final concentration of 50 µM DFBHI-1T. Next, secure the culture tube lid, and then invert culture tubes 3x-5x to mix the solution evenly before taking the first time point reading.
NOTE: The total percentage of DMSO within the culture tubes for E. coli should not exceed 10%, as this can affect cell viability16. - Remove the lid and place the culture tube into the sample tube lifter. Raise the holder by hand to the sample injection needle, and, under the proper sample file, click the Record icon. Additionally, begin a timer by pressing Start for the experiment.
- Lower the tube lifter by hand after the run is completed, and repeat steps 2.3.5.1-2.3.5.2. (with the timer running) by adding 1.4 µL of the concentrated DFHBI-1T, inverting the culture tubes, and taking readings for all the remaining biological replicates. These first recordings will be the readings obtained at 0 min for all the biological replicates. Do this one at a time for each biological replicate.
NOTE: Make a note of the time when the record flow cytometry acquisition is pressed. Adhere to this time staggering for time points over the course of the experiment. - Continue to take readings by raising the culture tubes in the sample tube lifter to the sample injection needle, selecting the proper sample file, clicking Record, and lowering the lifter by hand after each run is complete. Do this for all the additional time points and biological replicates being tested. Repeat the steps until the experiment is completed.
NOTE: Keep the samples out of light to avoid the photobleaching of DFHBI-1T in solution by covering the samples with aluminum foil.
- Add 1.4 µL of a concentrated dye stock (50 mM DFHBI-1T in DMSO) into 1x PBS solution with cells to give a final concentration of 50 µM DFBHI-1T. Next, secure the culture tube lid, and then invert culture tubes 3x-5x to mix the solution evenly before taking the first time point reading.
- Measure fluorescence at time points for cells without dye (Control).
- Run the appropriate cleaning protocols for the flow cytometer before repeating the experiment again with negative controls following the manufacturer's protocol. This is done to minimize any carryover from the previous experiment into the control time point analysis experiment. Below are the steps followed for the flow cytometer between experiments:
- Place an empty culture tube in the tube lifter by hand, raise the tube holder, and click on the Unclog icon in the Instrument tab. This will run a backflush in the fluidics system to clean up any sticky samples. Lower the tube holder by hand and remove the tube once the Unclog sequence is done.
- With a new culture tube, add 3 mL of a 10% bleach solution, place the culture tube into the tube holder, and raise the holder by hand to the sample injection needle. Additionally, place a clean 96-well plate into the autosampler if applicable to the flow cytometer.
- Click on the Sanitize SIP/Sanitize Autosampler SIP icon to run a cleaning sequence with 10% bleach throughout the fluidics system. Lower the tube holder to complete the cleaning sequence.
- Set the sample files for the control time point analysis run following the directions within step 2.3.3.
- In a new culture tube, prepare a dilute cell solution in 1.5 mL of 1x PBS solution. Add 3 µL of the induced cells in AI media into the 1x PBS solution to make a dilute cell solution. Repeat this step for each biological replicate.
- Add 1.4 µL of DMSO into the culture tube one at a time to the 1x PBS solution with cells and test the same time points. Secure the culture tube lid, and then invert culture tubes 3x-5x to mix the solution evenly before taking the first time point reading. Do this one at a time for each biological replicate.
- Follow the same protocol for the analysis of control cells as for cells with dye, using steps 2.3.5.2-2.3.5.4.
- Run the appropriate cleaning protocols for the flow cytometer before repeating the experiment again with negative controls following the manufacturer's protocol. This is done to minimize any carryover from the previous experiment into the control time point analysis experiment. Below are the steps followed for the flow cytometer between experiments:
- Shutdown the flow cytometer: Follow the manufacturer's protocol for the proper shutdown of the instrumentation. For the flow cytometer, the instrument is prepared for shutdown in the following manner:
- Perform the initial cleaning protocol for the flow cytometer following steps 2.3.6.1.1-2.3.6.1.3.
- Replace the culture tube with 10% bleach solution with a culture tube with 3 mL of focusing fluid. Raise the tube holder by hand, and, under the Shutdown icon, click on the drop-down menu and select Thorough.
NOTE: Optional pause point.
- Set up the flow cytometer.
- Analysis of the flow cytometry data
- Export all the FCS files for analysis. Open the FCS files with a flow cytometry analysis software.
- Using one of the Cell-only FCS files, generate a gate from the forward scatter (FSC) and side scatter (SSC) dot plot (FSC-Area/SSC-Area), using both log axes to exclude any signals from debris. To create this gate, click on the AutoGate icon on the flow cytometry analysis software and name it Gate 1. Apply this same gate to all the samples tested under the All Samples tab in the data processing workspace. This will result in Gate 1 underneath all the FCS files being processed.
- Create a new subset file with the Cell only FCS file used in step 2.4.2 by double-clicking it. Change the axis settings to FSC-Area/FSC-Height, with both using log axes. Click on the AutoGate icon on the flow cytometry analysis software to generate an oval gate, naming it Gate 2. Apply this gate as a subset underneath the gate set in step 2.4.2 to all the samples tested. This will result in all the samples having Gate 1 > Gate 2 associated with each FCS file.
- Create another subset file with both gates set in step 2.4.2 and step 2.4.3 applied by double-clicking Gate 2. Change the axis settings to FSC-Area/Histogram. Apply this histogram gate as a subset to all the samples tested, resulting in all the samples having Gate 1 > Gate 2 > Gate 2 associated with each FCS file.
NOTE: The histograms can be renamed from Gate 2 auf Histogram to assist with moving the histograms into the layout window, as well as to create more organization with the data processing. - To analyze the mean fluorescence intensity (MFI) values, open the layout window. Click and drag the histogram gates for each time point into the layout window.
- Perform a statistical analysis for "∑ Mean: BL1-A" (GFP) for each sample tested to display the MFI results on the layout window.
- Calculate the standard deviation for the MFI values per time point analysis for at least three independent biological replicates.
- Save the histograms and MFI values for each time point by exporting the layout window as a PDF file.
NOTE: Optional pause point.
- Graph flow cytometry data
- Open the PDF file containing the histograms and MFI values for each time point. The MFI values will be copied over into a data analysis software. In data graphing software, create a new data table in the XY format.
- Select to create an XY Table with the following selected:
Data Table: Enter or import data into a new table
Options:
X: Elapsed Times
Y: Enter (three to four) replicate values in side-by-side subcolumns - Label on the X axis all the time points for the experiment and control runs.
- In Group A, input the MFI values for all the biological replicates into each time point for fluorescence analysis of the cells with the dye added.
- In Group B, input the MFI values for all the biological replicates into each time point for analysis of the cells with no dye (DMSO) added.
- To observe the results, click on [Insert Data Set Name] under the Graphs tab. This will display the data points as means, with error bars representing the standard deviation (s.d.) at each time point. The X-axis represents the elapsed time, and the Y-axis represents the MFI values.
- Select to create an XY Table with the following selected:
- Open the PDF file containing the histograms and MFI values for each time point. The MFI values will be copied over into a data analysis software. In data graphing software, create a new data table in the XY format.
In vitro kinetics
The sequences of the DNA templates and primers, which are purchased as synthetic oligonucleotides, are shown in Table 2, and the reagent recipes are shown in Supplementary File 1. PCR amplification is used to scale up the amount of DNA template with the T7 promoter, which is required for the subsequent in vitro transcription (IVT) reaction. In addition, PCR amplification can be used for two purposes in the same reaction: to generate the full-length Broccoli DNA template by primer extension, as well as to scale up the DNA template.
After the IVT reaction to synthesize RNA, PAGE purification will remove any unwanted truncated transcripts, degraded products, and unreacted rNTPs from the full-length RNA product. This type of purification is preferred because truncated or degraded RNAs will cause the inaccurate determination of RNA concentrations. Since nucleotide bases absorb UV light, RNA bands and rNTPs on the gel can be visualized under UV as shadows against a fluorescent TLC plate. Thus, bands corresponding to the correct product size can be selectively extracted.
The nearest-neighbor method overestimates the extinction coefficients and, thus, the concentrations of structured RNAs since it does not account for hypochromicity due to base pairing17. Therefore, to determine accurate RNA concentrations, neutral pH thermal hydrolysis assays were performed to hydrolyze the RNA to individual NMPs18. The accurate extinction coefficient was calculated as a sum of the extinction coefficients of NMPs in the RNA sequence.
The kinetics of DFHBI binding to Spinach2 and Broccoli was determined using a plate reader assay. RNA was first renatured to ensure it would be in the correct conformation for dye binding. The reaction conditions for the plate reader kinetics assay consisted of 40 mM HEPES, pH 7.5 (KOH), 125 mM KCl, 3 mM MgCl2, 100 nM renatured RNA, and 10 µM DFHBI, and the reaction was measured at 37 °C. This temperature and concentration of MgCl2 were chosen to mimic physiological conditions19, though conditions of 28 °C and 10 mM MgCl2 may also be used for improved aptamer folding.
Both fluorogenic aptamers Spinach2 and Broccoli display two-phase association kinetics for binding to the DFHBI dye (Figure 2). The kinetics data were better fit by two-phase association than one-phase association for both aptamers (Supplementary Figure 1). The rate constants and t1/2 values for the fast and slow associations were determined by the best fit curve (Table 3). PercentFast, which describes what percent of the fluorescence turn-on magnitude is accounted for by the faster DFHBI-binding RNA population, was also determined.
Spinach2 in the binding-competent state shows faster turn-on than Broccoli (t1/2 = 1.2 s vs. 2.0 s). The second phase kinetics for both aptamers are similar (t1/2 = 180 s) and likely correspond to a common rate-limiting step for a sub-population of the sample (PercentFast = 68% and 60% for Spinach2 and Broccoli, respectively). Overall, these results show that well-folded Spinach2 and Broccoli aptamers exhibit very fast turn-on kinetics, with the initial half-maximal turn-on within 1-2 s of dye addition.
Cellular kinetics
The sequences of the DNA constructs, which are cloned into the pET31b plasmid, are shown in Table 2, and reagent recipes are shown in Supplementary File 1. The DNA constructs of fluorogenic RNA aptamers are typically contained within a tRNA scaffold for cellular experiments. The BL21 Star (DE3) E. coli strain is an expression strain with a mutation in RNase E that increases RNA stability.
The fluorescence time point measurements were recorded every 5 min for the first 45 min, followed by readings at 1 h, 1.5 h, and 2 h, giving a total of 12 time points plus the cell-only reading. Having the shortest time interval being 5 min permitted multiple biological replicates to be measured at each time point, with regular spacing between the replicate measurements of 30 s to 1 min. The total volume of cells diluted in 1x PBS solution used for the time course experiment was 1.5 mL.
The cells were gated prior to determining the mean fluorescence intensity (MFI) of the population of single cells. Gating selects an area on the scatter plot to determine the cell population that will be analyzed. This process prevents any debris or multiplet readings from being included in the analysis. For the flow cytometry analysis shown, 30,000 events were recorded, which resulted in 10,000-20,000 events analyzed after gating.
The cellular fluorescence kinetics are a function of both dye diffusion into E. coli and dye binding kinetics to the RNA aptamer within the cellular environment (Figure 1B). For cells expressing Spinach2-tRNA, it was observed that the mean fluorescence intensity (MFI) increases immediately at the "0" timepoint, due to the short time lag (in seconds) between dye addition and sample analysis (Figure 3A). Furthermore, cellular fluorescence has already reached its maximal equilibrated MFI value (40,441 ± 990) at the first time point of 5 min. In contrast, control cells show low background fluorescence (416) and no change in MFI value with DMSO addition (Figure 3B). A comparison between cells with dye and cells with no dye reveals that fluorescence activation is 98-fold ± 2 in cells. Overall, these results show that Spinach2-tRNA expressed in E. coli cells exhibits fast turn-on kinetics, with maximal turn-on within less than 5 min of dye addition.
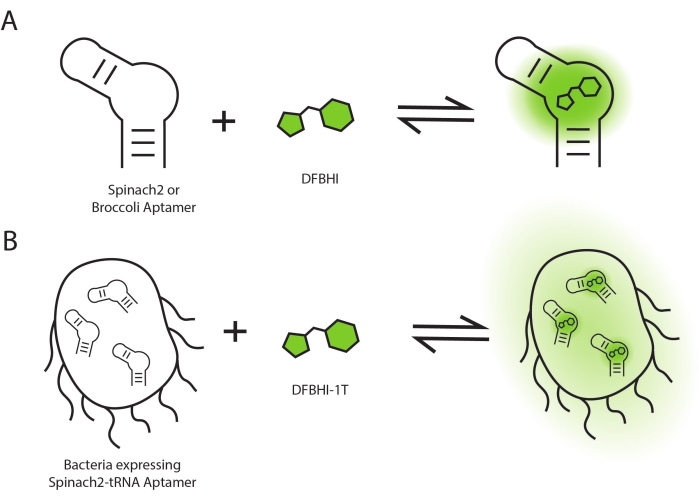
Figure 1: Schematic of fluorescence activation. Fluorescence activation occurs upon RNA aptamers binding to dye molecules (A) in vitro and (B) in cells. Please click here to view a larger version of this figure.
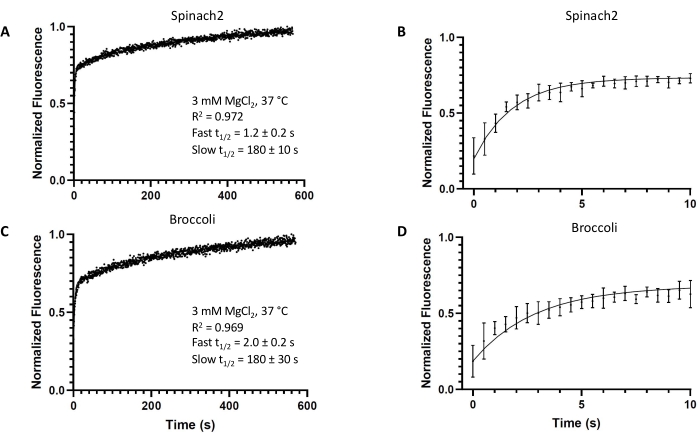
Figure 2: In vitro kinetics of the fluorogenic aptamers. Representative in vitro kinetics of the fluorogenic aptamers (A,B) Spinach2 or (C,D) Broccoli modeled by two-phase association, with t = 0 s being the timepoint of DFHBI addition (final DFHBI concentration: 10 µM). Experiments were performed in triplicate. All error bars represent standard deviations from the mean. From the fit, t1/2 values were obtained for both the fast and slow association reaction components. Please click here to view a larger version of this figure.
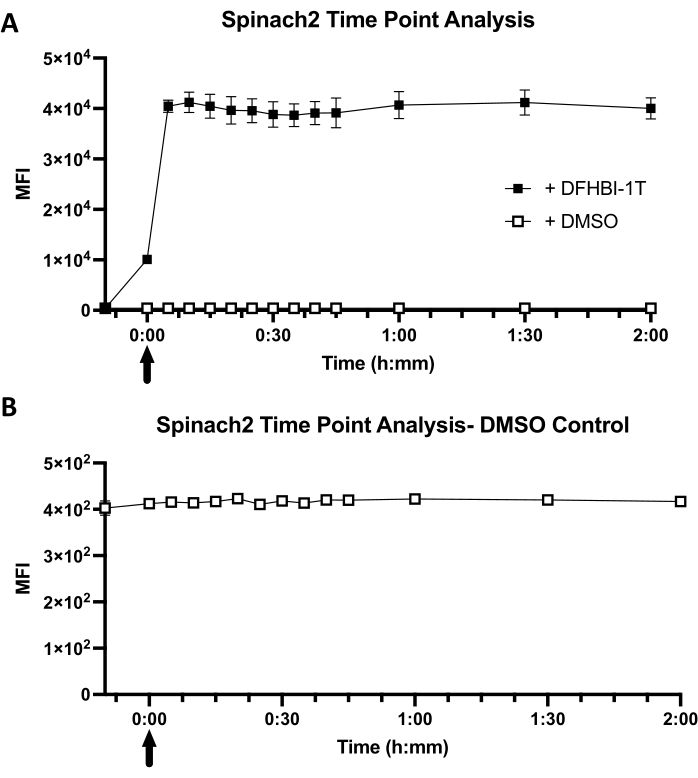
Figure 3: Representative cellular kinetics of Spinach2 in a tRNA scaffold. (A) Timepoint analysis of tRNA-Spinach2 dye uptake over the course of 2 h. A cell-only baseline was taken prior to adding in DFHIB-1T or DMSO. Time points were taken every 5 min for the first 45 min, followed by a time point reading at 1 h, 1.5 h, and 2 h. Arrow represents when the DFHBI-1T or DMSO was added into 1x PBS solution with BL21 Star cells. The final concentration of DFHBI-1T for analysis is 50 µM. For the DMSO control, the addition of DMSO was at an equal volume (1.4 µL) used for DFHBI-1T dye addition. (B) A close-up of the DMSO control time point analysis with BL21 Star E. coli cells. Mean fluorescence intensity (MFI) indicates the overall fluorescent readout of BL21 Star cells with dye or DMSO. Data represent the mean ± standard deviation of three biological replicates. Please click here to view a larger version of this figure.
| Aptamer-Dye Pair | Length (nt) | Max abs (nm) | Max em (nm) | Extinction coefficient (M-1·cm-1) | Quantum yield | Brightness | Kd (nm) | Tm (°C) | Reference |
| Spinach2-DFHBI | 95 | 445 | 501 | 26100 | 0.7 | 63 | 1450 | 37 | 4 |
| Spinach2-DFHBI-1T | 95 | 482 | 505 | 31000 | 0.94 | 100 | 560 | 37 | 13, 14 |
| Broccoli-DFHBI-1T | 49 | 472 | 507 | 29600 | 0.94 | 96 | 360 | 48 | 13 |
Table 1: Previously published photophysical and biochemical properties of Spinach2-DFHBI4, Spinach2-DFHBI-1T13,14, and Broccoli-DFHBI-1T13.
| Spinach2 + T7 promoter | 5’-CGATCCCGCGAAATTAATACGACTCACTATAGGATGTAACTGAATGAAATGGTGAA GGACGGGTCCAGTAGGCTGCTTCGGCAGCCTACTTGTTGAGTAGAGTGTGAGCTCC GTAACTAGTTACATC-3’ |
||
| Broccoli + T7 promoter | 5’-CGATCCCGCGAAATTAATACGACTCACTATAGgagacggtcgg gtccagatattcgtatctgtcgagtagagtgtgggctc-3’ |
||
| tRNA-Spinach2 construct (in pET31b plasmid) | 5'-CGATCCCGCGAAATTAATACGACTCACTATAGGGGCCCGGATAGCTCAGTCGGT AGAGCAGCGGCCGGATGTAACTGAATGAAATGGTGAAGGACGGGTCCAGTAGGCT GCTTCGGCAGCCTACTTGTTGAGTAGAGTGTGAGCTCCGTAACTAGTTACATCCGG CCGCGGGTCCAGGGTTCAAGTCCCTGTTCGGGCGCCA TAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTG-3' |
||
| Spinach2 Forward Primer | 5’-CGATCCCGCGAAATTAATACGACTCACTATAG-3’ | ||
| Spinach2 Reverse Primer | 5’-GATGTAACTAGTTACGGAGC-3’ | ||
| Broccoli Forward Primer | 5’-CGATCCCGCGAAATTAATACGACTCACTATAGgagacggtcgggtccagatattcgtatctg-3’ | ||
| Broccoli Reverse Primer | 5’-gagcccacactctactcgacagatacgaatatctggacccgaccgtctc-3’ | ||
Table 2: DNA sequence table containing DNA sequences and primers used for in vitro and cellular kinetics studies. Bold= T7 promoter; Underlined = tRNA scaffold; Caps = Spinach2; lowercase = Broccoli; Bold italics = T7 terminator.
| Aptamer | Fast t1/2 (s) | Slow t1/2 (s) | KFast (s-1) | KSlow (s-1) | Percent Fast |
| Spinach2 | 1.2 ± 0.2 | 180 ± 10 | 0.56 ± 0.07 | 0.0039 ± 0.0002 | 68 ± 5 |
| Broccoli | 2.0 ± 0.2 | 180 ± 30 | 0.35 ± 0.05 | 0.0039 ± 0.0006 | 60 ± 3 |
Table 3: In vitro kinetics values of the Spinach2 and Broccoli aptamers derived from fitted data. The data are reported as the mean ± standard deviation of three replicates.
Supplementary Figure 1: Representative in vitro kinetics of the fluorogenic aptamers. Representative in vitro kinetics of the fluorogenic aptamers (A,C) Spinach2 or (B,D) Broccoli modeled by one-phase association at (A,B) 600 s or (C,D) 20 s measurement times, with arrows indicating the timepoint of DFHBI addition (final DFHBI concentration: 10 µM). Experiments were performed in triplicate. Overall, these data are less well fit by a one-phase association model than a two-phase association model when fluorescence signal is monitored for a longer duration. Please click here to download this File.
Supplementary File 1: Recipes for in vitro kinetics experiment. Please click here to download this File.
