Protein Target Prediction and Validation of Small Molecule Compound
Summary
The experiment used here shows a method of molecular docking combined with cellular thermal shift assay to predict and validate the interaction between small molecules and protein targets.
Abstract
Proteins are fundamental to human physiology, with their targets being crucial in research and drug development. The identification and validation of crucial protein targets have become integral to drug development. Molecular docking is a computational tool widely utilized to investigate protein-ligand binding, especially in the context of drug and protein target interactions. For the experimental verification of the binding and to access the binding of the drug and its target directly, the cellular thermal shift assay (CETSA) method is used. This study aimed to integrate molecular docking with CETSA to predict and validate interactions between drugs and vital protein targets. Specifically, we predicted the interaction between xanthatin and Keap1 protein as well as its binding mode through molecular docking analysis, followed by verification of the interaction using the CETSA assay. Our results demonstrated that xanthatin could establish hydrogen bonds with specific amino acid residues of Keap1 protein and reduce the thermostability of Keap1 protein, indicating that xanthatin could directly interact with Keap1 protein.
Introduction
Proteins are highly important macromolecules in living organisms and possess a diverse range of unique functions within cells, such as membrane composition, cytoskeleton formation, enzyme activity, transportation, cell signaling, and involvement in both intracellular and extracellular mechanisms1,2,3. Proteins manifest their biological functions primarily through specific interactions with a variety of molecules, including other proteins, nucleic acids, small molecule ligands, and metal ions1,4. Ligands are small molecular compounds that specifically bind to proteins in an organism. The interaction between proteins and ligands occurs at specific sites on the protein, called the binding sites, also known as the binding pockets5. In medicinal chemistry research, the focus lies in identifying key proteins that are clearly associated with diseases, which serve as targets for drugs6. Therefore, gaining a deep understanding of the binding sites between proteins and ligands is of utmost importance in promoting drug discovery, design, and research7,8.
Molecular docking is a widely used computational tool for studying protein-ligand binding, which employs the three-dimensional structures of proteins and ligands to explore their primary binding modes and affinities when forming stable complexes9,10,11. The application of molecular docking technology originated in the 1970s. Based on the lock and key pairing principle and utilizing the algorithms of molecular docking software, one can determine the interaction between compounds and molecular targets by analyzing docking results. This approach enables the prediction of active binding sites for both the compound and the target molecule. Consequently, it facilitates the identification of an optimal binding conformation (here called the binding model) for ligand-receptor interactions, which is crucial for understanding the mechanics of these molecular engagements12,13,14,15. While molecular docking provides valuable computer-based predictions of ligand-receptor interactions, it is important to note that these are preliminary findings. Consequently, further experimental verification is essential to confirm these interactions.
The cellular thermal shift assay (CETSA), initially proposed by Pär Nordlund's research team in 2013, serves as a method for validating drug-target protein interactions. This technique specifically tests the thermal stability of target proteins induced by drug binding, providing a practical approach for confirming molecular interactions16,17,18. This approach is based on the fundamental principle that ligand binding initiates a thermal shift within target proteins and is applicable to a wide array of biological samples, including cell lysates, intact living cells, and tissues19,20. CETSA supports direct target engagement of small molecules in intact cells by detecting thermodynamic stabilization of proteins due to ligand binding and linking the observed phenotypic response to the target compound21,22. Among the various methodologies derived from CETSA, Western Blot-CETSA (WB-CETSA) is considered a classical approach. Following sample preparation using the CETSA method, western blot analysis is utilized to detect alterations in the thermal stability of the target protein. This allows for the precise determination of drug-protein interactions within cellular systems17,23.
Xanthatin is a bioactive compound isolated from the plant Xanthium L. with properties such as anti-inflammatory, which has been used in traditional Chinese medicine to treat diseases like nasal sinusitis and arthritis24,25. The kelch-like ECH-associated protein 1 (Keap1) is a component of the Cullin3-based Cullin-RING E3 ubiquitin ligase multi-subunit protein complex and an important regulator of intracellular redox homeostasis, which influences the intensity and duration of the inflammatory response by modulating the intracellular redox state26. In this study, we first utilized molecular docking to investigate the interaction between xanthatin (small molecule) and Keap1 protein, aiming to predict their binding mode. Subsequently, we employed the CETSA method to validate this interaction by assessing the impact of xanthatin on the thermal stability of the Keap1 protein.
Protocol
1. Downloading the structures of xanthatin and Keap1
- Open the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), input xanthatin (small molecule), then press Search and click on The First Result. Click on Download and click Save under 2D structure to save the compound in .sdf format.
- Download the crystal structure of the protein.
- Open the UniProt database (https://www.uniprot.org/), input Keap1 and click on Search. Click Human under popular organisms in the left column, and then click on The First Entry Named Q14145 in the right column.
- Click on Structure under the function in the left column, find the structure with PDB ID: 2FLU, and click on RCSB-PDB. Click on Download Files and select PDB Format to download the crystal structure of Keap1 protein.
2. Molecular docking
- Create a new folder named molecular docking on the desktop and save the downloaded xanthatin (small molecule) and Keap1 (protein) structures in this folder.
NOTE: Folder name and path must be in English. - Open Maestro software, click File, select Change Working Directory, click Desktop, double-click to select the newly created molecular docking folder and click Choose Options.
- Small molecule processing
- Click File and import structures options, click Desktop, double-click on molecular docking folder, click on xanthatin Structure File, and then click Open to import small molecule file.
- Click Tasks in the upper right corner of the software and select LigPrep option. Use structures from the workspace; the rest of the parameters are the default parameters and click Run to perform small molecule processing.
- Protein structure processing
- Click the File and Import Structures options, click Desktop, and double-click on the Molecular Docking folder. Click on the Keap1 Protein Structure file, and then click Open to import the protein structure file.
- Click Tasks in the upper right corner of the software and select the Protein Preparation Wizard option. Check the box Before Deleting Waters Beyond 5 Å From Het Groups and enter a pH of 7.4+/-0.0. Click Preprocess, click Refine, and then click Optimize > Remove Waters > Minimize.
- Click on the next task when the previous task is completed. Click Tasks and select the Sitemap option, use default settings for all parameters, and click Run.
- Click File and Import Structures options, click Desktop, double-click on the molecular docking folder, and open the folder named sitemap_1. Click on the first file named Sitemap_1_out.maegz, and then click Open.
- Double-click on the dot to select sitemap_1_protein in the workspace, and then click on Sitemap_1_site_1 to select it . Click Tasks and select the Receptor Grid Generation option.
NOTE: The protein sites predicted by the sitemap are ranked according to the scoring, with the highest-scoring site in the first place. - Select the Entry option under receptor, with the remaining parameters unchanged. Click on the Small White Ball in the protein structure to generate a docking box of default size and change the job name to glide-grid_2FLU. Click Run.
- Molecular docking
- Click Tasks and select the Ligand Docking option. Select the Receptor Grid As from the file and click Durchsuchen. Click Desktop, double click on molecular docking folder, double click on glide-grid_2FLU file, click Glide-grid_2FLU.zip file, and then click Open.
- Click on Use Ligands From Files > Browse > Desktop. Double-click on the molecular docking folder, double-click on the ligprep_1 file, click Ligprep_1-out.maegz file, and then click Open.
- Click Settings and select Precision as XP (extra precision) option, change the job name to glide-dock_XP_2FLU, and click Run.
- View docking results
- Click File and Import Structures options, click Desktop, and double-click on the Molecular Docking folder. Double-click on the glide-dock_XP_2FLU file, click the glide-dock_XP_1_pv.maegz file and then click Open.
- Double-click on the dot to select ligand in the workspace, and then click to select sitemap_1_protein. Click Table, slide to the far right, and view the score under docking score.
- Visualization of 2D results
- Double click on the dot to select ligand in the workspace, and then click to also select sitemap_1_protein. Click Ligand Interaction > View and check the LID legend box.
- Click File, choose Save Screenshot, input 6000 at the width, uncheck the box for Transparent Background, and click OK. Click on Desktop, name the file as 2D-xanthatin-2FLU, and save the image.
- Visualization of 3D results
- Double click on the dot to select ligand in the workspace, and then click to also select sitemap_1_protein.
- Click L in Quick Select to select small molecules, and then click Style. Click on the Second Line in the style to change the size of the small molecule bond and click on the color atoms to change the small molecule color.
- Select the small molecule in the figure, click the right mouse button, select Expand Selection, and choose 4 Å. Click Apply Labels in style to display the labels.
- Click on the second line in the style to change the size of amino acid bonds and click on the Color Atoms to change amino acid color.
- Click on Next > Invert, hold down the control key and click on the first eye position under style to show only the amino acids with labels.
- Click on Style > Ribbons to show the protein as a ribbon style and click on Edit Ribbon under ribbons to change the ribbon color.
- Click on Interactions Toggle in the bottom right corner of the screen, click the right mouse button and uncheck contacts/clashes.
- Search for measure in tasks, select Distance, and click on the two atoms connected by the yellow dotted line (hydrogen bond) in the screen to measure hydrogen bonds lengths.
- Press and hold the mouse's wheel to rotate the protein, adjust the protein to the center of the screen, and ensure that each amino acid label is visible.
- Click Workspace > Save Image as and click on the desktop. Save the image as 600dpi by default, name the file as 3D-xanthatin-2FLU, choose file type as tiff format, and save the image.
3. Cell culture and CETSA sample preparation
- Cell culture
- Add 5 x 106 RAW264.7 cells/mL into a 100 mm culture dish and incubate with 6 mL of DMEM medium with 10% FBS and 1% antibiotic-antimycotic solution at 37 °C, 95% humidity, and 5% CO2. Prepare three dishes and perform the experiment when the cells reach 80% confluency.
- CETSA sample preparation
- Aspirate the old medium, add 2 mL of phosphate buffer saline (PBS) warmed to room temperature to each dish, and wash 2x.
- Add 2 mL of PBS to each dish of cells, mix the cells, and collect cells into two 1.5 mL microcentrifuge tubes. Centrifuge at 377 x g for 3 min at room temperature.
- Discard the supernatant and resuspend cells with 2 mL of PBS containing 1% Broad-spectrum protease inhibitor mixtures. Divide into individual microcentrifuge tubes with 1 mL of cell suspension in each.
- Freeze microcentrifuge tubes in liquid nitrogen until white solid, then immediately thaw in a 37 °C water bath and repeat 3x. Centrifuge at 12,000 x g at 4 °C for 10 min.
- Take two microcentrifuge tubes, one with 100 µM xanthatin and one with an equal volume of DMSO, and add 450 µL of centrifuged supernatant to each tube, incubate at 37 °C for 30 min.
- Add the supernatant from the two microcentrifuge tubes to 14 PCR tubes at a volume of 60 µL/tube, and heat tubes simultaneously at varying temperatures (45 °C, 48 °C, 51 °C, 54 °C, 57 °C, 60 °C and 63 °C) for 3 min, with two PCR tubes at each temperature, for the DMSO and xanthatin groups, respectively.
- For the unheated group, take supernatant from each of the two microcentrifuge tubes and add to 14 PCR tubes, 7 for the DMSO group and 7 for the xanthatin group, in a volume of 60 µL each. Do not heat the samples here.
- Cool the tubes at room temperature for the heated group. Centrifuge all the tubes (heated and unheated) at 12,000 x g for 10 min at 4 °C and take 48 µL of supernatant from each tube to a new 1.5 mL microcentrifuge tube.Add 12 µL of SDS-PAGE protein loading buffer to each tube, vortex to mix well, and heat in a water bath at 100 °C for 5 min.
NOTE: Prepared CETSA samples must be stored at -80 °C and tested by western blot within 1 week.
4. Western blot
- Preparation of SDS-PAGE gel
- Rinse one long and one short glass plate with ultrapure water. Place the plates into the clamping slot, then on a transparent board. Add ultrapure water and leak check for 10 min.
- Add 8 mL of lower gel solution and 8 mL of lower gel buffer into a conical flask and mix by gently shaking. Add 160 µL of coagulant promoter to the gel solution and mix well.
NOTE: The volume of the lower layer of adhesive should not exceed the green line on the clamping slot. - Drain the ultrapure water from the plates and blot them dry with filter paper. Add the prepared lower gel solution. Add 2 mL of isopropyl alcohol and wait for 20 min for solidification.
- Take 2 mL of upper gel solution and 2 mL of upper gel buffer into a conical flask and mix by gently shaking the flask.
- Remove the isopropyl alcohol from the gel layer and blot the excess isopropyl alcohol with filter paper.
- Add 40 µL of coagulant promoter to the mixed upper gel solution, mix well, then add between the long and short glass plates and insert a 1.5 mm 15-hole comb, wait for 20 min.
NOTE: The comb should be inserted perpendicular to the upper layer, with no air bubbles between the comb and the solution.
- Electrophoresis
- Add 1 L of ultrapure water to a beaker, add a sachet of SDS-PAGE running buffer powder, stir well with a glass rod, and add the prepared gel to the electrophoresis unit. Place the unit in the box and pull out the comb.
- Thaw marker and CETSA samples at room temperature, vortex, and spin down. Add 2.5 µL of marker to the first well and 20 µL of CETSA samples to each of the remaining wells according to the predetermined sequence.
NOTE: CETSA samples were added successively according to temperature treatment described in step 3.2.6 from 45 °C to 63 °C; the DMSO group was added first, then the xanthatin group (Figure 1). - Add the remaining electrophoresis solution to the electrophoresis unit, close the lid, and insert the electrode, positive pole to positive pole, negative pole to negative pole.
- Turn ON the power switch and adjust the voltage switch to 80 V to start electrophoresis.
- Transfer to membrane
- Add 850 mL of ultrapure water, 100 mL of anhydrous ethanol, and 50 mL of rapid transfer buffer to a beaker and stir well. Prepare foam board, plastic knives, tweezers, trays, and membrane transfer devices.
- Cut the PVDF membrane according to the size of the gel, mark it, and soak it in methanol for 30 s to activate it.
- Pour the transfer solution into the tray, place and open the transfer clamps, and place a sponge pad and three layers of transfer filter paper.
- Stop electrophoresis and then open the lid of the electrophoresis box. Take the electrophoresis device, pour out the electrophoresis solution, and remove the plate. Place the plate on the foam board, gently pry it off with a plastic knife, and cut the gel to clearly visualize the protein marker and the target proteins.
- Hold one side of the marker with tweezers, wash it in a tray containing the transmembrane solution, and lay it flat on filter paper. Dip the activated PVDF membrane into the transfer solution, hold the labeled side with tweezers, and cover the gel face-down.
NOTE: The PVDF membrane marked side is the front side and pay attention to the fact that no bubbles are generated during the placement process. - Place two layers of transfer filter paper and one layer of sponge pad on the PVDF membrane, cover and clamp the transfer clips, and put them into the transfer tank.
NOTE: Black color of transfer clip to black color of transfer slot. - Add the remaining transmembrane solution to the transmembrane cassette, cover with the lid, positive to positive, negative to negative.
- Adjust the current to 400 mA, time to 30 min, and then press Run to start transferring the membrane at room temperature.
- Blocking
- Add 2 L of ultrapure water to a glass vial, add a sachet of TBS powder, and then add 2 mL of Tween to make TBST solution. Weigh the BSA powder to prepare a 5% BSA solution with TBST and add 6 mL of BSA solution to the incubation box.
- Stop the membrane transfer and then open the transfer clip. Clamp the marker side of the PVDF membrane and place it in the incubation box. Place the incubation box on a shaker and block for 1.5 h at room temperature.
- Incubation of primary antibody
- Prepare the primary antibody (see Table of Materials for details) with 5% BSA solution at 1:1000, recycle the BSA solution in the incubator box, and add 6 mL of primary antibody to the incubation box. Place the incubation box in a foam box with ice packs and keep it on a shaker overnight.
- Incubation of secondary antibody
- Collect the primary antibody in the incubation box the next day, then add 6 mL of TBST solution and place it on a shaker to wash for 5 min.
- Pour off the washed TBST solution and add fresh for washing; repeat 5x and prepare the secondary antibody with 5% BSA solution at 1:5000.
- Pour off the TBST solution and add 6 mL of secondary antibody to the incubation box. Place the incubation box on a shaker and incubate for 1.5 h at room temperature.
- Collect the secondary antibody (see Table of Materials for details), then add 6 mL of TBST solution and place it on a shaker to wash for 5 min. Pour off the washed TBST solution and add fresh for washing; repeat 5x.
- Exposing the strips
- Retain the TBST solution at the end of the last wash. Take equal volumes of chemiluminescent reagents A and B, shake, and mix well. Shield the solution from light.
- Open the gel imaging system, click on Samples, check calibration, wait for calibration to complete, and then click on Marker, check calibration.
- Pick up the marker side of the strip with tweezers and fully immerse in the chemiluminescent reagent solution. Clamp one side of the marker to place the strip face down in the imager, close the lid of the imager, click on Exposure, and set the exposure time to 1 s.
NOTE: The developer must completely cover the strip, and the development should be in a dark environment. - Click Save, name the image, and save it as a .tif file. Analyze the optical density of the western blot. The gray values of each temperature in the DMSO group were compared with the gray values of 45 °C, and the gray values of each temperature in the xanthatin group were compared with the gray values of xanthatin at 45 °C.
Representative Results
Molecular docking analysis predicted the interaction between xanthatin and Keap1 protein. Figure 2 demonstrates the formation of hydrogen bonds between xanthatin and amino acid residues Gly-367 and Val-606 of Keap1 protein, with a hydrogen bond length of 2.17 Å for Gly-367 and 2.13 Å for Val-606. In addition, the calculated docking score of -5.69 kcal/mol signifies a good binding affinity between xanthatin and Keap1 protein.
The CETSA method showed that xanthatin binding shifted the thermal stability of Keap1 protein, confirming the interaction between xanthatin and Keap1 protein21. Figure 3 indicates that xanthatin remarkably shifts the thermal stability of Keap1 protein within the temperature range of 48 °C to 57 °C, as compared to the DMSO group. To ensure equal sample loading, firstly, the density of cell inoculation in which we performed the experiments was consistent; secondly, the cell samples from the DMSO group and the xanthatin group were mixed well in the cell culture dishes and then were equally distributed into two microcentrifuge tubes, which ensured the consistent amount of cell samples, and, the volume of supernatant added to each PCR tube was 60 µL, and lastly, the volume of each sample uploaded in the Western blot was 20 µL, which ensured equal loading for heated samples. Protein degradation in samples from around 51 °C is mainly due to changes in protein structure and accelerated chemical reactions caused by high temperatures. The above results demonstrate that xanthatin could directly bind to Keap1 protein.

Figure 1: Loading order of western blot. The temperature in order from left to right were 45 °C, 48 °C, 51 °C, 54 °C, 57 °C, 60 °C, and 63 °C. The first lane was added with a marker; two lanes were used for each temperature; the first lane was the DMSO group, and the second lane was the xanthatin group. Please click here to view a larger version of this figure.
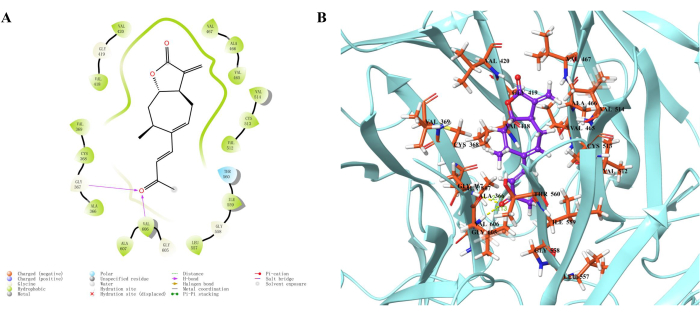
Figure 2: Interaction between xanthatin and Keap1 (PDB: 2FLU) protein. (A) The 2D representation of xanthatin binding to Keap1. (B) The 3D visualization of xanthatin and Keap1 protein. Please click here to view a larger version of this figure.
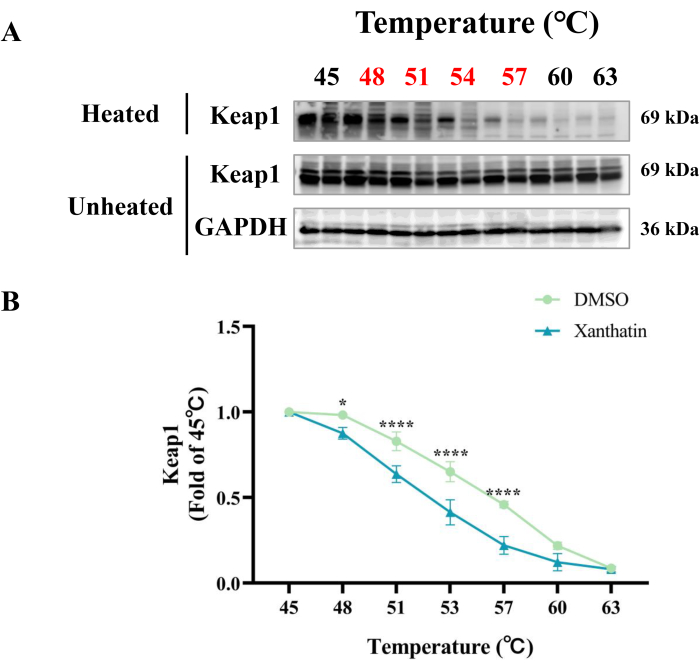
Figure 3: Effect of xanthatin on the thermal stability of Keap1 protein. (A) The effects of xanthatin on the thermal stability of Keap1 were detected by CETSA. (B) The optical densities of Keap1 were normalized to those obtained at 45 °C. Data are represented as mean ± SD. Statistical analysis was performed by one-way analysis of variance (one-way ANOVA). n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Please click here to view a larger version of this figure.
Discussion
The identification of disease targets and the discovery and development of drugs are closely interconnected27. By precisely targeting specific targets, drug candidates can be developed to treat particular diseases more effectively while concurrently minimizing the side effects associated with the drugs28,29. The most commonly used targets are protein targets30. However, the identification of special protein targets poses an immense challenge due to the extensive diversity of proteins within cells30,31. In this paper, we adopted an approach that combines molecular docking with CETSA to identify and validate protein targets. The results revealed a direct interaction between xanthatin and Keap1 protein. The integration of computer technology and biological experiments can also be employed in the investigation of other drugs and protein targets to ascertain the presence of a direct interaction between the drug and the target. This approach, which combines prediction with verification, effectively reduces both time consumption and economic costs. Furthermore, the experimental instruments and materials are easy to obtain, and the operation process is not complicated. Finally, The CETSA samples are easy to prepare and test by the western blot method, which ensures the reproducibility of the samples.
In our experience, there are several critical aspects that require special attention during experimental procedures. Firstly, it is essential to select the crystal structure of the protein resolved by X-ray analysis when choosing the protein structure before molecular docking. Secondly, cell density and freezing/thawing time are crucial factors in CETSA experiments. Cells in the logarithmic growth phase should be selected for CETSA experiments as their growth state is stable and has minimal impact on experimental results. During repeated freezing and thawing of cells, timing should be well-controlled with liquid nitrogen used for freezing until a white solid form, followed by immediate thawing at room temperature before subsequent freezing cycles. Lastly, western blot experiments must be completed promptly after preparing CETSA samples to prevent protein degradation.
Molecular docking and CETSA are both well-established techniques that play a crucial role in the field of drug development. Molecular docking is instrumental in predicting molecular-level interactions between compounds and their targets, offering valuable insights into potential binding modes10,11. CETSA, in contrast, focuses on assessing the impact of drugs on protein thermal stability and serves as a tool for validating drug-protein interactions17. While CETSA is a relatively straightforward method for verification, it is also a low throughput way by using western blot for the study of thermal shift of drug-protein complex, and it should not be the sole technique relied upon. In addition, the steps of CETSA are complicated, and any step errors during the operation can have a great impact on the results. Furthermore, the CETSA method cannot determine the specific binding site and binding mode of the compound to the protein target and cannot indicate whether the binding is covalent or non-covalent17,18,23. Beyond thermal shift assay, other kinds of energetic assay, such as pH-dependent protein precipitation (pHDPP), can also be used for the study of ligand-protein interactions32. Moreover, thermal proteome profiling is an energy-based method for revealing ligand-protein interactions, allowing high-throughput, large-scale analysis of protein-drug interactions by combining proteomics with CETSA33.
In addition, localized surface plasmon resonance (LSPR) can provide further qualitative and quantitative analysis34,35. It not only determines the existence of interactions between a drug and its target but also measures the affinity parameters and kinetic parameters of these intermolecular interactions, offering a more comprehensive understanding.
Beyond thermal shift assay, pH-dependent protein precipitation (pHDPP) can also be used for the study of ligand-protein interactions32. Molecular docking is a physico-mechanics-based simulation method for predicting interactions between small molecules and biomolecules. This method aims to achieve binding by optimizing the conformation and orientation of small molecules to produce the best complementarity between them and biomolecules. Target prediction utilizes artificial intelligence techniques to predict the binding sites between small molecules and biomolecules. The two approaches have key differences in method application and prediction accuracy. Method application: molecular docking is mainly used for drug design and optimization, providing a theoretical basis for new drug discovery. Target prediction, with the help of more advanced artificial intelligence, is more often applied to high-throughput screening and rapid discovery of lead compounds to improve the efficiency of drug discovery. Prediction accuracy: Since molecular docking is based on physical simulations, its prediction accuracy is higher, but the computational cost is also relatively high. While target prediction has a lower computational cost, the prediction accuracy depends on the abundance of training data and model selection.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
This work was supported by National Natural Science Foundation of China (82004031) and Sichuan Science and Technology Program (2022NSFSC1303). We express our great appreciation to Jiayi Sun at Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, for the assistance with western blot.
Materials
| 0.45 μm Polyvinylidene fluoride membrane | Millipore | PR05509 | |
| Anhydrous ethanol | Chron chemicals | 64-17-5 | |
| Bovine serum albumin | BioFroxx | 4240GR100 | |
| Broad-spectrum protease inhibitor mixtures | Boster Biological Technology Co., Ltd | AR1193 | |
| DMSO | Boster Biological Technology Co., Ltd | PYG0040 | |
| Enhanced chemiluminescence reagent | Beyotime Biotechnology Co., Ltd | P0018S | |
| GAPDH antibody | ProteinTech Group Co., Ltd | 10494-1-AP | |
| Gel Imaging Instrument | E-BLOT | Touch Imager Pro | |
| Gradient PCR instrument | Biometra TADVANCED | Biometra Tadvanced 96SG | |
| High-speed freezing centrifuge | Beckman Coulter | Allegra X-30R | |
| Horseradish peroxidase-conjugated affiniPure goat antibody | ProteinTech Group Co., Ltd | SA00001-2 | |
| Isopropyl alcohol | Chron chemicals | 67-63-0 | |
| Keap1 antibody | Zen BioScience Co., Ltd | R26935 | |
| Metal bath | Analytik Jena | TSC | |
| Methanol | Chron chemicals | 67-56-1 | |
| Ncmblot rapid transfer buffer (20×) | NCM Biotech Co., Ltd | WB4600 | |
| Omni-Easy OneStep PAGE gel fast preparation kie | Epizyme Biotech Co., Ltd | PG212 | |
| Phosphate buffer saline | Boster Biological Technology Co., Ltd | PYG0021 | |
| Prestained Color Protein Marker | Biosharp | BL741A | |
| Protein Blotting Electrophoresis System | Bio-Rad | MiniPROTEANÒTetra Cell | |
| RAW264.7 cell | Beyotime Biotechnology Co., Ltd | C7505 | |
| RAW264.7 cell-specific medium | Procell Life Science&Technology Co., Ltd | CM-0597 | |
| SDS-PAGE protein loading buffer | Boster Biological Technology Co., Ltd | AR1112-10 | |
| SDS-PAGE running buffer powder | Servicebio | G2018 | |
| Tris buffered saline powder | Servicebio | G0001 | |
| Tween 20 | BioFroxx | 1247ML100 | |
| Water bath | Memmert | WNE10 | |
| Water purifier | Millipore | Milli- IQ 7005 | |
| Xanthatin | ChemConst Biotechnology Co., Ltd | CONST210706 |
Referenzen
- Soleymani, F., Paquet, E., Viktor, H., Michalowski, W., Spinello, D. Protein-protein interaction prediction with deep learning: A comprehensive review. Computat Struct Biotechnol J. 20, 5316-5341 (2022).
- Du, X., et al. Insights into protein-ligand interactions: mechanisms, models, and methods. Int J Mol Sci. 17 (2), 144 (2016).
- Wu, Q., Peng, Z., Zhang, Y., Yang, J. COACH-D: improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 46, W438-W442 (2018).
- Zhang, F., et al. PROBselect: accurate prediction of protein-binding residues from proteins sequences via dynamic predictor selection. Bioinfo. 36, i735-i744 (2020).
- Kandel, J., Tayara, H., Chong, K. T. PUResNet: prediction of protein-ligand binding sites using deep residual neural network. J Cheminfo. 13 (1), 65 (2021).
- Dhakal, A., Mckay, C., Tanner, J. J., Cheng, J. Artificial intelligence in the prediction of protein-ligand interactions: recent advances and future directions. Brief Bioinform. 23 (1), 476 (2022).
- Hatty, C. R., Banati, R. B. Protein-ligand and membrane-ligand interactions in pharmacology: the case of the translocator protein (TSPO). Pharmacol Res. 100, 58-63 (2015).
- Chaires, J. B. Calorimetry and thermodynamics in drug design. Ann Rev Biophys. 37, 135-151 (2008).
- Huang, S. Y., Zou, X. Advances and challenges in protein-ligand docking. Int J Mo Sci. 11 (8), 3016-3034 (2010).
- Li, J., Fu, A., Zhang, L. An overview of scoring functions used for protein-ligand interactions in molecular docking. Interdiscip Sci. 11 (2), 320-328 (2019).
- Crampon, K., Giorkallos, A., Deldossi, M., Baud, S., Steffenel, L. A. Machine-learning methods for ligand-protein molecular docking. Drug Discov Today. 27 (1), 151-164 (2022).
- Pinzi, L., Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int J Mol Sci. 20 (18), 4331 (2019).
- Abdolmaleki, A., Ghasemi, F., Ghasemi, J. B. Computer-aided drug design to explore cyclodextrin therapeutics and biomedical applications. Chem Biol Drug Des. 89 (2), 257-268 (2017).
- Chen, G., Seukep, A. J., Guo, M. Recent advances in molecular docking for the research and discovery of potential marine drugs. Mar Drugs. 18 (11), 545 (2020).
- Li, T., Guo, R., Zong, Q., Ling, G. Application of molecular docking in elaborating molecular mechanisms and interactions of supramolecular cyclodextrin. Carbohydr Polym. 276, 118644 (2022).
- Martinez Molina, D., et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 341 (6141), 84-87 (2013).
- Tu, Y., Tan, L., Tao, H., Li, Y., Liu, H. CETSA and thermal proteome profiling strategies for target identification and drug discovery of natural products. Phytomedicine. 116, 154862 (2023).
- Dai, L., et al. Horizontal cell biology: Monitoring global changes of protein interaction states with the proteome-wide cellular thermal shift assay (CETSA). Annu Rev Biochem. 88, 383-408 (2019).
- Lade, D. M., Nicoletti, R., Mersch, J., Agazie, Y. M. Design and synthesis of improved active-site SHP2 inhibitors with anti-breast cancer cell effects. Eur J Med Chem. 247, 115017 (2023).
- Jiang, X., et al. Novel chemical-structure TPOR agonist, TMEA, promotes megakaryocytes differentiation and thrombopoiesis via mTOR and ERK signalings. Phytomedicine. 110, 154637 (2023).
- Mateus, A., et al. Thermal proteome profiling for interrogating protein interactions. Mol Syst Biol. 16 (3), 9232 (2020).
- Sanchez, T. W., et al. Real-time cellular thermal shift assay to monitor target engagement. ACS Chem Biol. 17 (9), 2471-2482 (2022).
- Tolvanen, T. A. Current advances in CETSA. Front Mol Biosci. 9, 866764 (2022).
- Liu, M., et al. Xanthatin inhibits STAT3 and NF-κB signalling by covalently binding to JAK and IKK kinases. J Cell Mol Med. 23 (6), 4301-4312 (2019).
- Liu, Y., Chen, W., Zheng, F., Yu, H., Wei, K. Xanthatin alleviates LPS-Induced inflammatory response in RAW264.7 macrophages by Inhibiting NF-κB, MAPK and STATs activation. Molecules. 27 (14), 4603 (2022).
- Dinkova-Kostova, A. T., Kostov, R. V., Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys. 617, 84-93 (2017).
- Tabana, Y., Babu, D., Fahlman, R., Siraki, A. G., Barakat, K. Target identification of small molecules: An overview of the current applications in drug discovery. BMC Biotechnol. 23 (1), 44 (2023).
- Schenone, M., Dančík, V., Wagner, B. K., Clemons, P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 9 (4), 232-240 (2013).
- Hughes, J. P., Rees, S., Kalindjian, S. B., Philpott, K. L. Principles of early drug discovery. Br J Pharmacol. 162 (6), 1239-1249 (2011).
- Chakraborty, C., et al. SARS-CoV-2 protein drug targets landscape: A potential pharmacological insight view for the new drug development. Expert Rev Clin Pharmacol. 14 (2), 225-238 (2021).
- Ziegler, S., Pries, V., Hedberg, C., Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew Chem Int Ed Engl. 52 (10), 2744-2792 (2013).
- Zhang, X., et al. Highly effective identification of drug targets at the proteome level by pH-dependent protein precipitation. Chem Sci. 13 (42), 12403-12418 (2022).
- Zhang, X., et al. A simplified thermal proteome profiling approach to screen protein targets of a ligand. Proteomics. 20, 1900372 (2020).
- Hou, Y., et al. Salidroside intensifies mitochondrial function of CoCl2-damaged HT22 cells by stimulating PI3K-AKT-MAPK signaling pathway. Phytomedicine. 109, 154568 (2022).
- Wang, X., et al. Salidroside, a phenyl ethanol glycoside from Rhodiolacrenulata, orchestrates hypoxic mitochondrial dynamics homeostasis by stimulating Sirt1/p53/Drp1 signaling. J Ethnopharmacol. 293, 115278 (2022).
