ATG9A is a transmembrane protein associated with several intracellular membrane compartments8,17,22,23,24. In basal conditions, ATG9A is mainly localized at the trans-Golgi network (TGN), as indicated by the immunofluorescence of the endogenous protein and the overlaps with GM130, a cis-Golgi marker (Figure 1A), as well as in small vesicles that partially overlap with the endocytic recycling compartment (ERC)23. ATG9A localization at the Golgi can be detected using different immunofluorescence protocols. However, the vesicular fraction of ATG9A, as well as its change of localization, in particular the increase in the vesicular pool, in response to specific stimuli such as nutrient and serum starvation, can be quite variable in intensity and difficult to visualize with conventional imaging approaches. The ratio between ATG9A localized at the Golgi and ATG9A localized to a vesicular fraction is termed the ATG9A dispersal rate. To detect changes in the ATG9A dispersal rate, for instance upon EBSS treatment, which is used to deplete both serum and amino acids, a Golgi marker such as GM130 or TGN46 and a cytoskeleton marker such as Phalloidin, which stains the cell contour25, are useful to readily quantify the ATG9A dispersal (Figure 1B). Importantly, the mean fluorescence ratio analysis can only be interpreted as a comparative measure between conditions rather than as a fixed rate of dispersal. The ratio between compartments is highly dependent on biological and non-biological factors such as the cell line used, the staining quality, or the thresholding methods applied (Figure 1B). For this reason, the researcher needs to set up a pipeline that is able to detect ATG9A Golgi enrichment in their specific experimental conditions and then extend the analysis with the same parameters to all the images in the set to be analyzed. Representative binary images and areas selected for the analysis of ATG9A mean fluorescence are shown as a guide in Figure 1B.
ATG9A harbors several transmembrane domains flanked by two relatively flexible and unstructured N- and C-terminal domains, of which the C-terminal sequence encompasses almost half the protein12. Importantly, the localization pattern of overexpressed ATG9A can be influenced by which protein end is tagged (Figure 2A). In particular, when using transient expression systems and tagging ATG9A directly on its N-terminus with a fluorescent tag (e.g., eGFP, mRFP, or derivatives), its Golgi localization can be partially compromised, with less enrichment seen in basal (i.e., fed) conditions, while the ATG9A vesicles are still readily visible (Figure 2A). Tagging ATG9A on its C-terminus seems to slightly induce larger GFP positive clusters that could be aggregated. Finally, a monomeric version of mRFP-ATG9A also shows similar fluorescent clusters of vesicles and little Golgi staining in overexpressing cells (Figure 2A).
ATG9A folds in the ER membrane before being trafficked to the Golgi and ATG9A vesicles. During its residence in the ER, ATG9A becomes modified by N-linked glycans on Asparagine 99, and then upon reaching the Golgi, it acquires complex, mature N-linked glycans1,14. This modification by glycosylation can be detected through western blot by the appearance of a double band14. Consistent with its intracellular localization, most endogenous ATG9A harbors complex N-linked glycans, and, therefore, the higher-molecular weight band is predominant, with a faint lower-molecular weight band also visible (Figure 2B). The presence of a double band is most readily seen when using Tris-acetate gels to improve the resolution of higher-molecular weight proteins (Figure 2B, control, t = 0). When the endogenous protein is subjected to PNGase F (Peptide:N-glycosidase F) treatment, which removes most of the complex N-linked glycans, the protein runs as a single band (Figure 2B, PNGase F, t = 0). Therefore, the N-linked glycosylation status of ATG9A can be used as a proxy to monitor the exiting of ATG9A from the ER to the Golgi, which is reflected by the relative ratio between the two bands.
When transfecting mRFP-ATG9A constructs transiently, the overexpressed protein initially accumulates in the ER, potentially because the trafficking machinery is unable to fold and traffic all the ATG9A, and the lower molecular weight band is predominant (Figure 2C, control t = 0). Notably, after 24 h of expression of mRFP-ATG9A, there is approximately an equal distribution between the upper and lower bands, suggesting that the mRFP-ATG9A pool is moving into the Golgi (Figure 2C, Control, t = 24). If the cells are treated with cycloheximide (CHX), which blocks de novo protein synthesis26, the folding and exit of ATG9A from the ER can be clarified. As the endogenous protein is folded, glycosylated, and resident in the Golgi, treatment with CHX does not significantly alter the ratio of the lower- and higher-molecular weight bands (Figure 2B, Control). However, using the transient expression of mRFP-ATG9A, the CHX treatment promotes the accumulation of the higher-molecular weight band (Figure 2C, Control, CHX t = 24). The higher-molecular weight overexpressed mRFP-ATG9A band collapses into the lower band after treatment with PNGase F (Figure 2C, PNGase F, t = 24). These data show that the endogenous protein rapidly acquires mature glycans, as reflected by the predominance of the higher-molecular weight band, and the CHX chase does not affect the ratio of the double bands (Figure 2B). In the case of transiently overexpressed mRFP-ATG9A, CHX treatment induces the accumulation of the upper band, indicating that more mature glycans are acquired as the ER pool folds and exits the ER to the Golgi (Figure 2C).
The addition of a linker between the ATG9A sequence and the fluorescent tags can be helpful in promoting a more physiological localization and trafficking of the protein. Fusing a 3x-FLAG sequence (24 amino acids) between an N-terminal fluorophore and ATG9A helps the overexpressed protein behave similarly to the endogenous one (Figure 3). Indeed, overexpressed mCherry-3xFLAG-ATG9A colocalizes with the Golgi marker GM130 in fed conditions (Figure 3A). Importantly, this localization and the ATG9A vesicular compartment are preserved over time, allowing the spatiotemporal study of the trafficking of ATG9A (Figure 3B).
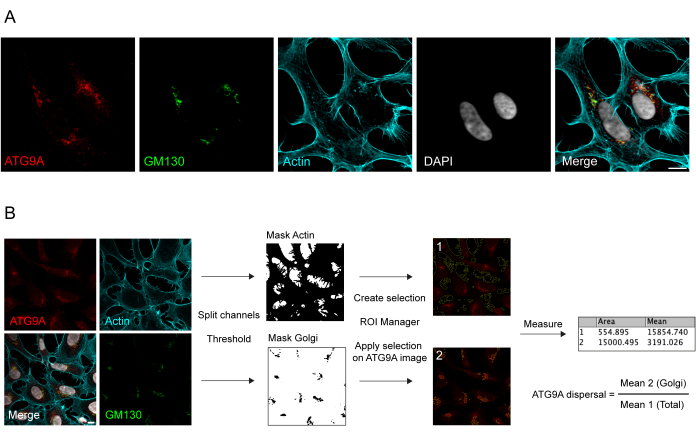
Figure 1: Image analysis of endogenous ATG9A localization. (A) Representative immunofluorescence image of endogenous ATG9A (red), GM130 as a Golgi marker (green), and Phalloidin to visualize the actin cytoskeleton (cyan). Scale bar = 10 μm. (B) Workflow of the image analysis to determine the fraction of endogenous ATG9A that localizes at the Golgi area. Scale bar = 10 μm. Please click here to view a larger version of this figure.
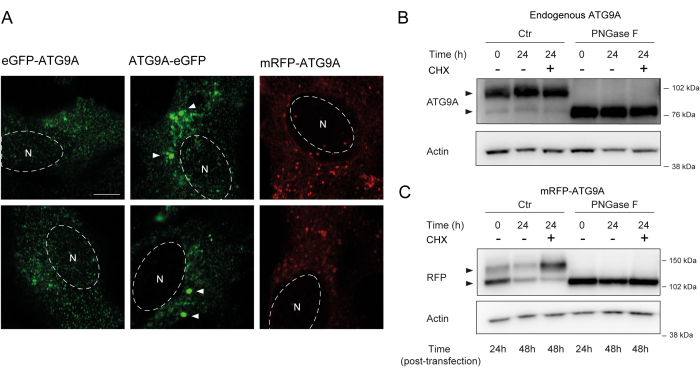
Figure 2: Analysis of fluorescently tagged-ATG9A constructs by localization and glycosylation. (A) eGFP N-terminally tagged ATG9A is less localized at the Golgi and primarily resides in the vesicles. eGFP C-terminally tagged ATG9A exhibits aggregates within the cell (some examples are marked by white arrowheads; the eGFP-ATG9A and ATG9A-eGFP are in green). mRFP N-terminally tagged ATG9A is less localized at the Golgi and primarily resides in the vesicles. N denotes the approximate location of the cell nucleus, and the mRFP-ATG9A is in red. Scale bar = 5 μm. (B) Endogenous ATG9A appears as two bands when analyzed by western blot (arrowheads): an upper band (complex N-linked glycans) and a lower band (no mature N-linked glycans). Treatment with cyclohexamide (CHX) does not affect the ratio between the upper and lower bands. Treatment with PNGase F causes the disappearance of the upper band. (C) After transient transfection of mRFP-tagged ATG9A in HEK293A cells, two prominent bands are visible on western blot (arrowheads). Treatment with PNGase F causes the disappearance of the upper band. Treatment with CHX after transfection leads to increased glycosylation as the pool of transfected ATG9A is trafficked from the ER to the Golgi. Please click here to view a larger version of this figure.
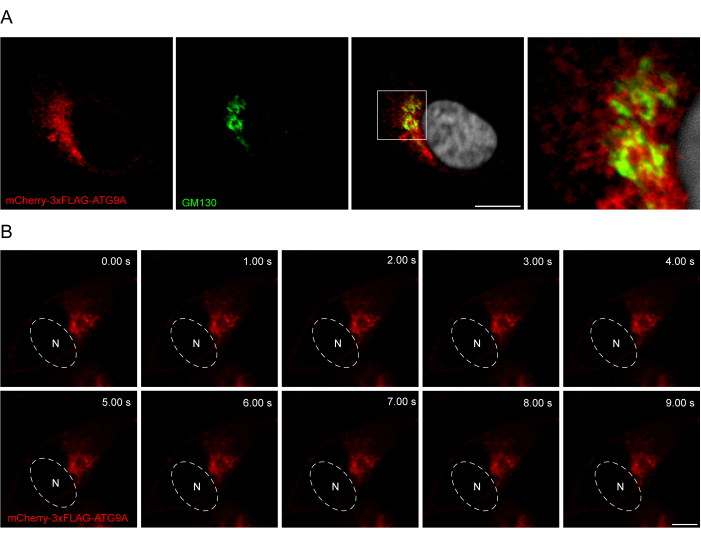
Figure 3: Analysis of mCherry-3xFLAG-ATG9A localization by immunofluorescence and live imaging. (A) Immunofluorescence experiments of HEK293A cells transiently overexpressing mCherry-3xFLAG-ATG9A and stained with the Golgi marker GM130. Scale bar = 10 μm. The mCherry-3xFLAG-ATG9A is in red, and the GM130 Golgi marker is in green. (B) Montage from live-imaging experiments in HEK293A cells transiently overexpressing mCherry-3xFLAG-ATG9A. N denotes the approximate location of the nucleus. Time frame = 1 fps. Scale bar = 10 μm. The mCherry-3xFLAG-ATG9A is in red. Please click here to view a larger version of this figure.
