1. Prepare a list of names for genes that you believe are co-regulated for analysis by SCOPE.
Save the list as a text file or copy it to the clipboard to paste into SCOPE in step 3. The file should contain one gene name per line with no additional information. Alternatively, you can prepare the list as a FASTA file containing the actual sequences to be analyzed.
2. Start your web browser and connect to the URL: http://genie.dartmouth.edu/SCOPE/
3. Enter the information that SCOPE needs to perform the analysis.
The initial SCOPE page is shown in Figure 1. Different sections are addressed in this step.
- Use the ‘Species’ popup menu to choose the species you will be examining. It is important to choose the correct species because SCOPE refers to the genome to calculate background frequencies of occurrence for any candidate motif it is examining.
- Use the ‘upstream sequence” radio buttons to choose either intergenic or fixed length. Intergenic will analyze all the sequence between of the gene you are looking at and the previous (upstream) gene. This will mean that different upstream lengths will be used for each gene. Choosing fixed length will look at exactly that number of nucleotides upstream from the start of the current gene. In this case, SCOPE will examine the same length of upstream sequence for each gene, even if that extends into the previous gene (or not). Typically, 800 nts is the best length to choose, but this can vary with species.
- Next tell SCOPE what gene set to analyze either by pasting in your gene list into the gene list text box, or by pressing the ‘choose file’ button to select the file containing the list of genes that you created earlier. You may, alternatively, paste in a FASTA sequence file into the same text box.
- The next section of the page contains a checkbox for ‘Examine genome for other genes containing found motif(s)?’ This option can add considerable analysis time since SCOPE has to evaluate every other gene in the genome. However, this can be very useful in identifying other genes that are good candidates for being co-regulated with the genes in the starting gene set. Since SCOPE analyses are relatively quick, it is suggested that you leave this off in your initial analysis. It can always be turned on from the results page to rerun the analysis, as explained in the results section.
- The ‘Results must include’ section can be used to enter a motif that you want SCOPE to include in its analysis. You might want to do this if you are looking for a specific motif.
- The last section on the page can be used to enter your email address and a comment to be saved with the analysis. If this is filled in, SCOPE will send an email with a link back to the web page containing results, and it will also include two attachments. One is a plain text file that has all the analysis results in human readable format. The second attachment contains an XML file that has every result that SCOPE has found in a computer readable format. If you want to do some additional analysis on the results, the XML file is very useful. Both files are “zipped” before being sent with the email.
- For this demo, we will start with the same information. This can be easily achieved by pressing the ‘Sample Search’ button which will fill in the necessary information. Press this button now. Three genes will be entered for you and appropriate choices made for the other fields. Leave these as they are set. The three genes are involved in telomere maintenance in Saccharomyces cerevisiae. The filled in form is shown in Figure 2. Press the ‘Run SCOPE’ button at the bottom of the page to start the analysis.
4. Representative Results:
The main results of the analysis are shown in Figure 3. The top of the page contains a table of information about the motifs that were found by SCOPE. The first column contains a list of motifs that were found and small colored squares serve as a legend for the graphical motif map shown below. The display of any given motif may be toggled on or off by clicking in the colored box (or where the colored box would be). This can be very useful to hide the display of highly repeated motifs that might make it difficult to see the less prevalent motif patterns.
Other columns of data are Count (the number of occurrences of that motif in the entire gene set), Sig value (an indication of the significance of that motif), Coverage (the percentage of the submitted genes that contain at least one instance of that motif), and Algorithm (which of the three component algorithms was used to detect the motif).
Clicking on any of the listed motifs will take the user to a page containing detailed information for that motif. The results details are shown for the cyan motif (atgnnnnttg) in Figure 4. On this page, the motif is represented in three ways: a sequence logo, a position weight matrix, and a list of all motif instances with their positions, strands and genes.
A little further down the page are some additional details about the results of looking for other genes containing this motif. As can be seen, in this case there were 1344 other genes containing the motif, all of which actually improved the Sig value when added to the original gene set. Pressing ‘Add checked genes to search’ will return to the SCOPE setup page with these genes added to the original gene set and the parameters set as they were previously. In this case, 10 extra genes are added to the original three.
Figure 5 shows the results of the analysis containing the extra genes for this motif. The original three genes are on the bottom of the results (in lower case). Looking at the pattern of motifs in the upstream region of these extra genes clearly shows that they are similar. In fact, many of these genes are involved in telomere maintenance as were the original three genes. Note also that the original motif is now the highest scoring motif in this set.
Another set of SCOPE results is shown in Figure 6. In this case, the set of genes are those that are involved in ribosome biogenesis in Saccharomyces cerevisiae. These genes are not actually part of the ribosome but are responsible for assembling ribosomes and include a number of modification enzymes. What is clear in the figure is that the red and green motifs form a reliable pattern that is likely to be involved in regulation of the genes in this set. We are investigating this pattern of “modules” in more detail and will report on it in a later publication.
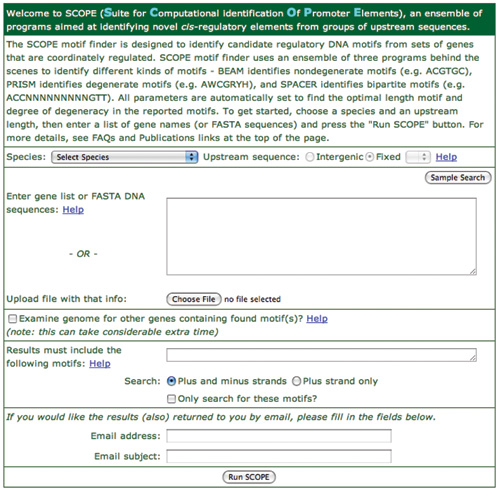
Figure 1. Main SCOPE input page. This page is used to enter the genes to be analyzed and to define the species and the length of upstream region to be examined. Optionally, the user can request the results by email or restrict the search to any specified motif. Video help is also available.
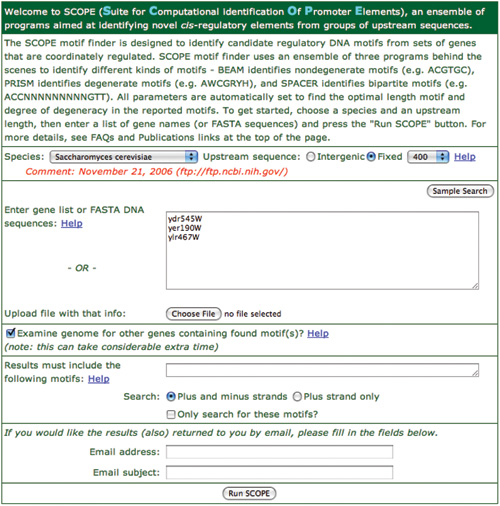
Figure 2. Main SCOPE input page with values filled in for performing a search. These parameters are the result of pressing the ‘Sample Search’ button. In this case, the check box to find other genes containing the motifs found by SCOPE is checked. This option takes longer to compute (every gene in the genome has to be examined) but can provide interesting insights.
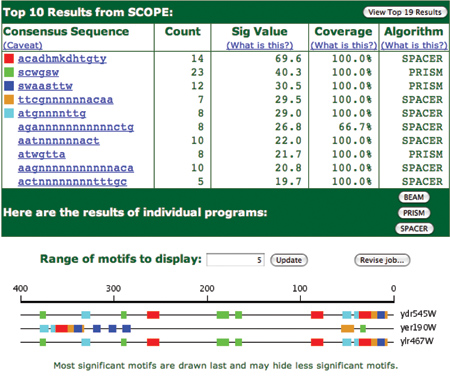
Figure 3. Main SCOPE results page. This page summarizes the results of the SCOPE search. A list of all high scoring motifs is provided and a color coded motif map shows the positioning of the identified motifs in the set of genes analyzed. Clicking on a colored box next to a motif will toggle the display of that motif on or off in the motif map. In addition to a significance score (Sig value), the fraction of genes containing the motif (coverage), and the algorithm used to find that motif are also provided.
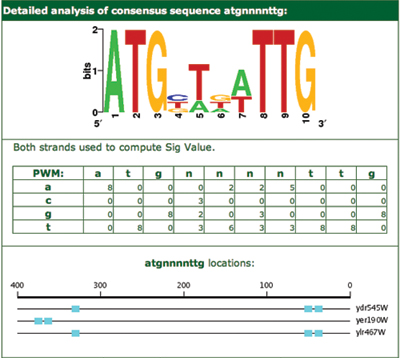
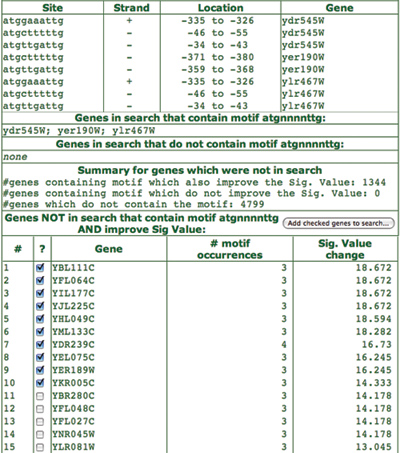
Figure 4. This results detail page is brought up when a specific motif is clicked in the main results page. It shows details of the individual motif. The sequence logo, the position weight matrix, and the consensus sequence each represent a different kind of summary of the list of motif instances also on the page. Since ‘find extra genes’ was checked in the original search setup, there is also information on this page about any other genes in the genome that contain this motif. From this page it is also possible to start another SCOPE run including the extra genes identified on this page.
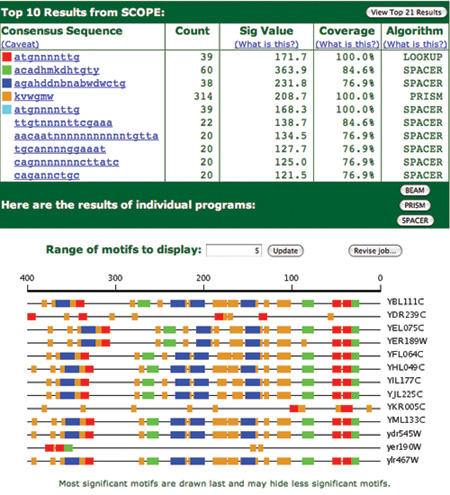
Figure 5. This figure shows the results of looking for extra genes for the motif ‘atgnnnnttg’ shown in Figure 4. The original three genes are in lower case at the bottom of the motif map. The additional genes are shown in upper case. There is a clear pattern to the motifs in the upstream regions of these genes. Notice also that the specified motif shows an algorithm as ‘LOOKUP’ because that is how it was identified. It actually matches the 5th motif found by SPACER in this analysis.
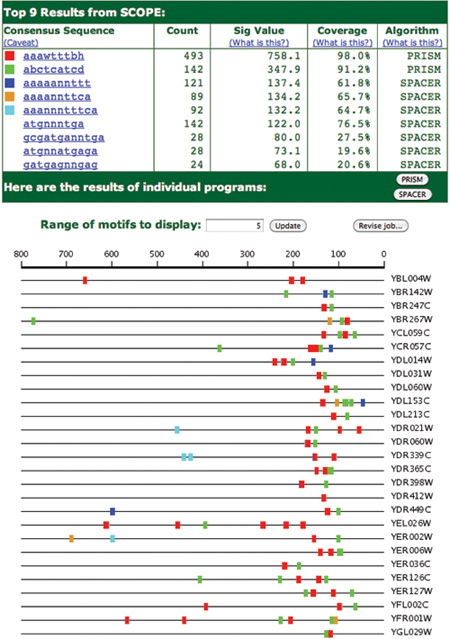
Figure 6. SCOPE output for genes involved in ribosome biogenesis in Saccharomyces cerevisiae. Note the conserved pattern of modules consisting of the motifs ‘aaawtttbh’ (red) and ‘abctcatcd’ (green) separated by about 10-30 nts and present at 100-200 nucleotides upstream of transcription start for the gene.
