In the early postnatal cerebellum, GCs exhibit significant changes in their mode and speed of migration as they cross different cortical layers1 (Figure 1). This section illustrates examples of results that can be obtained by studying GC migration in their natural cellular milieu. P10 rat cerebellar tissue slices labeled with a green fluorescent dye are examined under a confocal macroscope (Figure 3A) and we show that GCs migrate radially in the ML with an average speed of 18 µm/hr (Figure 3B, C). To date, the role of interactions/communications between neuronal and glial cells including the regulatory factors and molecular mechanisms involved in the control of cell migration in each cortical layer are largely unknown. Consequently, the main issue is to identify neuropeptides, neurotransmitters, neurotrophins and extracellular matrix components that could play a role in these cortical layer-specific changes of the speed during their migration process. Pituitary adenylate cyclase-activating polypeptide (PACAP) is detected mainly in the PCL, but also in the ML and the IGL during the first two postnatal weeks in rodents7,10,11. Application of PACAP38 (10-6 M) to the culture medium resulted in a 79% speed decrease of the GC in the ML. For example, the migration velocity of GCs in the ML dropped from 11.9 µm/hr in control conditions to 2.5 µm/hr after administration of PACAP38 (Figure 4A). Tissue-type plasminogen activator (tPA) is a member of the proteolytic cascade that leads to the degradation of the extracellular matrix (EM) components such as cell adhesion molecules or laminin12,13. tPA and plasminogen, a substrate of tPA, are detected in cortical layers during development of the postnatal cerebellum14,15,16. Administration of PAI-1 (10-7 M), an inhibitor of endogenous tPA, reduced by 78% the GC migration in the ML. For example, GCs reduced migration speed in the ML from 19.2 µm/hr in control conditions to 4.2 µm/hr after addition of PAI-1 (Figure 4B). These results indicate that PACAP exerts a direct inhibitory effect on GC movements and that the serine protease tPA facilitates the migration of GCs in the ML of the developing rat cerebellum.
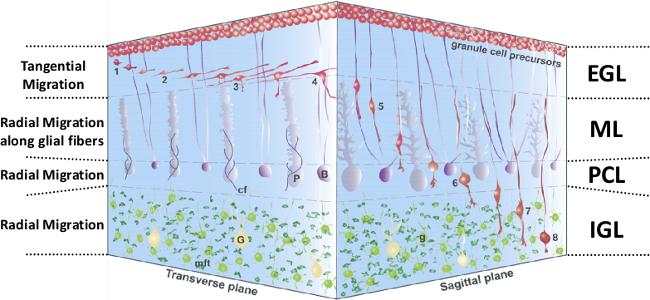
Figure 1: 3D representation of GC migration in the postnatal cerebellar cortex. 1-4, Extension of GC processes and tangential migration in the EGL. 5, Radial migration in the ML along Bergmann glial fibers. 6, Transient stationary phase in the PCL. 7, Glial-independent radial migration in the IGL. 8, Completion of GC migration in the IGL. GC, granule cell, in red; EGL, external granular layer; B, Bergmann glia, in dark purple; G, Golgi cell, in yellow; cf, climbing fibers, in blue; g, postmigratory granule cells, in light green; IGL, internal granular layer; mft, mossy fiber terminal, in dark green; ML, molecular layer; P, Purkinje cell, in light purple; PCL, Purkinje cell layer. This figure has been modified from 5.
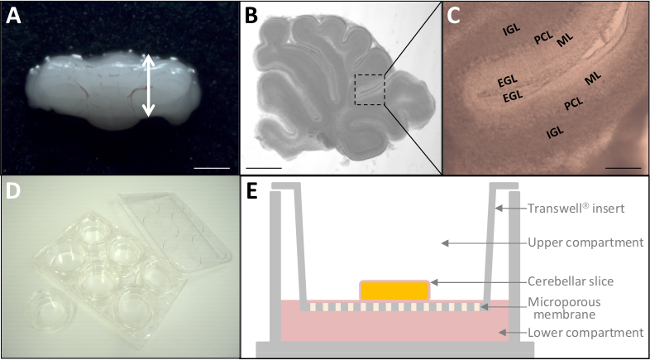
Figure 2: Ex vivo culture of P10 cerebellar slices. (A) Dissected cerebellum from P10 rat. Scale bar = 6 mm. (B) Micrograph of living 180 µm-thick cerebellar slice through stereomicroscopy. Scale bar = 3 mm. (C) At a higher magnification, the four cortical layers (EGL, ML, PCL, IGL) of the cerebellum are already distinguishable. Scale bar = 1 mm. (D) After fluorescent labeling, tissue slices are placed on culture inserts (24 mm diameter) into a 6-well plate. (E) Schematic representation of a culture insert with tissue culture treated polyester membrane. Please click here to view a larger version of this figure.
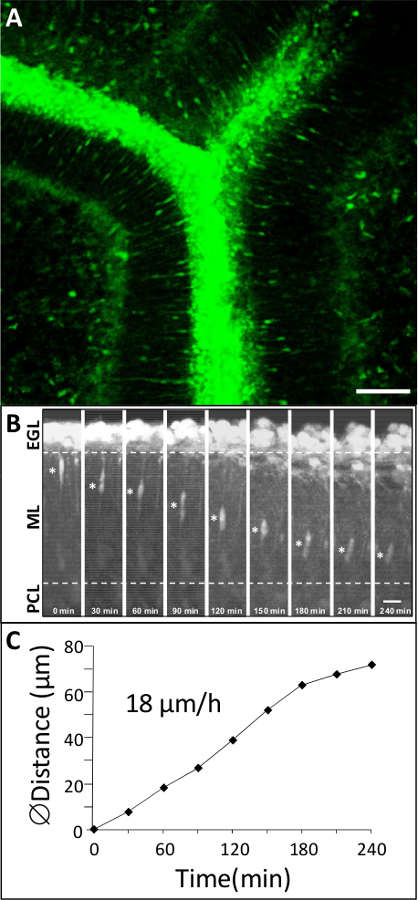
Figure 3: Dynamic migration of GCs in the cortical layers of rat P10 cerebellum. (A) Macroconfocal view (xyz, 2D projection) of a P10 rat cerebellar slice in which GCs are labeled with a green cytoplasmic fluorescent dye. Scale bar = 75 µm. (B) Time-lapse imaging showing GC movements in the ML by confocal macroscopy for 4 hr in control conditions. Asterisk (*) symbol marks the GC soma. Elapsed time (in min) is indicated on the bottom of each photomicrograph. Scale bar = 10 µm. (C) Sequential changes in the distance traveled by GC soma.
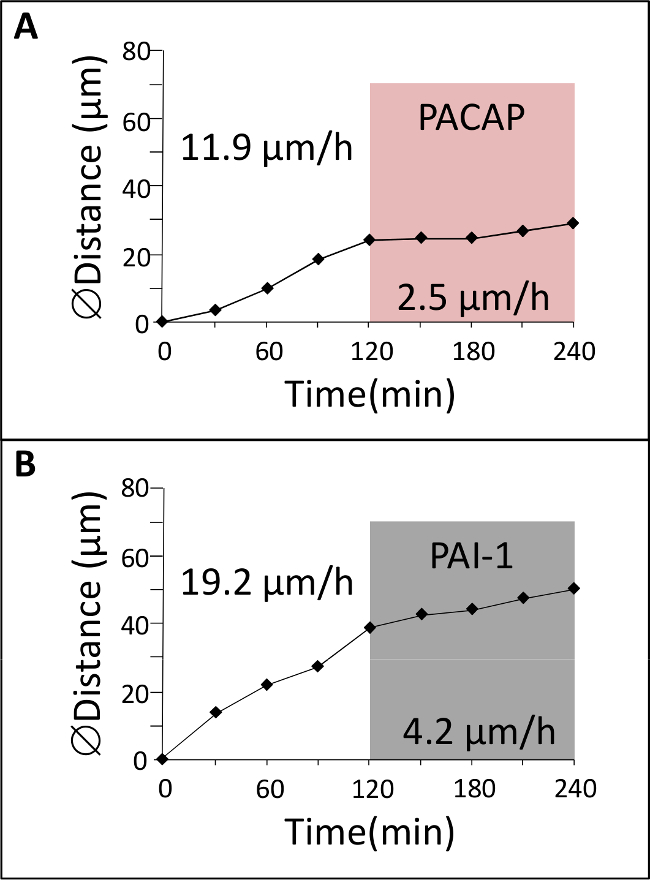
Figure 4: Effect of neuropeptide and protease inhibitor of GC migration. (A) GC was tracked in the ML by confocal macroscopy for 2 hr in control conditions and then for 2 hr in the presence of Pituitary adenylate cyclase-activating polypeptide (PACAP). (B) GC was tracked in the ML by confocal macroscopy for 2 hr in control conditions and then for 2 hr in the presence of plasminogen activator inhibitor-1 (PAI-1).
