1. MLV Transduction of Human Cells
- Isolate target cells and transduce them with an mLV-derived retroviral vector harboring the eGFP reporter gene and pseudotyped with Vesicular Stomatitis Virus G (VSV-G) or the amphotropic envelope glycoprotein16.
- Keep mock-transduced cells as a negative control for the following analyses. Since mLV-based retroviral vectors can transduce efficiently dividing cells, culture the target cell population in conditions that stimulate cell division. Transduction conditions need to be specifically optimized for each cell type under study. Cell growth and transduction conditions for human hematopoietic progenitors, T cells and epidermal cells are described in References 7, 14, 15, and 17.
- 48 h after transduction, resuspend 100,000 cells in 300 μL of phosphate-buffered saline (PBS) containing 2% fetal bovine serum and measure eGFP expression by flow cytometric analysis (488-nm excitation laser). Use mock-transduced cells as negative control. For optimal integration site retrieval, purify GFP+ cells by Fluorescence Activated Cell Sorting (FACS).
- Collect 0.5 to 5 million cells for genomic DNA preparation. A longer culture period (>7 days) is preferred to dilute the unintegrated provirus and necessary when analyzing the long-term progeny of bona fide stem cells (see Discussion Section and reference15). The cell pellets can be snap-frozen and stored at -80 °C until use.
2. Amplification of mLV integration sites by linker-mediated-PCR (LM-PCR)
- Preparation of genomic DNA (gDNA)
- Extract gDNA using a column-based DNA extraction kit and follow the protocol for cultured cells, according to the manufacturer's instructions.
- Restriction enzyme digestion
- Set up 4 restriction enzyme digestions per sample in 1.5 mL tubes. Digest 0.1 to 1 μg gDNA in each tube by adding 1 µL of Tru9I (10U) and 1 µL of Buffer M in a final volume of 10 µL. Incubate the reactions at 65 °C for 6 h or overnight.
- Add to each reaction 1 µL of PstI (10U), 1 µL of Buffer H and 8 µL of water. Incubate the reactions at 37 °C for 6 h or overnight. Samples can be stored at -20 °C until use.
- Linker ligation
- Prepare a 100 μM Tru9I linker stock solution in a 1.5 mL tube by mixing the linker plus strand and linker minus strand oligonucleotides at a 100 μM concentration. Put the tube in a water set at 100 °C and let it cool down at room temperature. Tru9I linker stock solution can be stored at -20 °C until use.
- Set up 8 linker ligation reactions in 1.5 mL tubes. For each reaction, add the following components to 10 µL of restriction enzyme digestion: 1.4 µL of 10X T4 DNA Ligase Reaction buffer, 1 µL of 10 μM linker Tru9I, 1 µL (2,000U) of T4 DNA ligase and 0.6 µL of water. Incubate at 16 °C for 3 to 6 h. Samples can be stored at -20 °C until use.
- First PCR
- Set up 48 PCR reactions (6 reactions from each ligation tube) in 0.2 mL tubes. For each PCR, add the following reagents to 2 µL of ligation reaction: 5 µL of 10X PCR buffer, 2 µL of 50 mM Magnesium Sulfate, 1 µL of 10 mM deoxynucleotide (dNTP) Mix, 1 µL of 10 μM linker primer, 1 µL of 10 μM mLV-3' LTR primer, 0.3 µL (1.5U) of Taq DNA Polymerase and 37.7 µL of water. Primer sequences are provided in Table 1.
- Perform PCR reaction in a thermal cycler with heated lid, as follows: 95 °C for 2 min; 25 cycles of 95 °C for 15 s, 55 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min; hold at 4 °C. Samples can be stored at -20 °C until use.
- Second PCR
- Set up 48 PCR reactions (1 from each "first PCR" tube) in 0.2 mL tubes. For each PCR, add the following components to 2 µL of the first PCR reaction: 5 µL of 10X PCR buffer, 2 µL of 50 mM Magnesium Sulfate, 1 µL of 10 mM dNTP Mix, 1 µL of 10 μM linker nested primer, 1 µL of 10 μM mLV-3' LTR nested primer, 0.3 µL of Taq DNA Polymerase (1.5 U) and 37.7 µL of water. Use nested primers designed for the specific massive sequencing strategy chosen: (i) linker nested primer and mLV-3' LTR nested primer compatible with Illumina platform; (ii) linker nested primer and mLV-3' LTR nested primer compatible with Roche platform. Primer sequences are listed in Table 1.
- Perform PCR reaction in a thermal cycler with heated lid, as follows: 95 °C for 2 min; 25 cycles of 95 °C for 15 s, 58 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min; hold at 4 °C. Samples can be stored at -20 °C until use.
- Pool the 48 nested PCR reactions in a 15 mL tube (final volume of ~2.4 mL). Samples can be stored at -20 °C until use.
- Determine the presence and the size of the LM-PCR products by agarose gel electrophoresis.
- Add 4 µL of Loading Buffer to 20 µL of LM-PCR products and load the sample onto a 1% agarose gel, together with a 100 bp DNA ladder. Run the gel at 5 V/cm for 30 to 60 min and visualize the PCR products by ethidium bromide staining.
- Run an LM-PCR reaction from the mock-transduced sample as negative control.
- Precipitate the amplicons by adding 0.1 volumes of sodium acetate solution (3 M; pH 5.2) and 2.5 volumes of 100% ethanol. Mix and freeze at -80 °C for 20 min.
- Spin at full speed in a standard microcentrifuge at 4 °C for 20 min. Discard the supernatant and wash the pellet with 70% ethanol. Spin at full speed in a standard microcentrifuge at 4 °C for 5 min.
- Discard the supernatant and air dry the pellet. Add 200 µL of PCR-grade water and resuspend the DNA. Samples can be stored at -20 °C until use.
- Add 20 μL of Loading Buffer to 100 µL of the LM-PCR products and load the sample onto a 1% agarose gel, together with a 100 bp DNA ladder.
- Run the gel at 5 V/cm for 30 to 60 min and cut the portion of the gel containing 150 to 500 bp-long amplicons.
- Purify the LM-PCR products with a column-based gel extraction kit and measure the concentration using an UV spectrophotometer.
- Use 1 µL of library to evaluate the length of the LM-PCR products using a bioanalyzer instrument, according to manufacturer's instructions.
- Shotgun cloning of amplicons
- Use 20 ng of the purified LM-PCR products and clone them in the pCR2.1-TOPO vector, according to the manufacturer's instructions.
- Perform sequencing reactions using the M13 Universal primer, followed by genomic mapping of the resulting sequences, to reveal the presence of the viral-genome junctions (including the mLV-3' LTR nested primer) in >50% of the clones to identify samples suitable for massive sequencing. Example of a viral-genome junction:

MLV-3' LTR nested primer 454 and linker nested primer 454 (Table 1) are indicated in bold and the 3' end of the viral LTR in italic. The human genomic sequence is highlighted in red text (chr10:6439408-6439509, hg19).
3. Massive Sequencing of mLV Integration Sites
NOTE: LM-PCR products can be sequenced using commercial platforms (choosing the proper nested primer pair in the second PCR reaction, see subsection 2.5.1). For sequencing by Roche GS-FLX pyrosequencing platform, refer to previous papers7,14,15. In this section, a newly-optimized protocol for Illumina sequencing platform is described.
- Library preparation
- Set up 1 indexing PCR reaction per sample in 0.2 mL tubes. For each PCR, add the following reagents to 5 µL (150-170 ng) of purified LM-PCR product: 5 µL of Index Primer 1, 5 µL of Index Primer 2, 25 µL of 2X Master Mix and 10 µL of PCR-grade water. Use a different index combination for each sample.
- Perform PCR reactions in a thermal cycler with heated lid, as follows: 95 °C for 3 min; 8 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s; 72 °C for 5 min; hold at 4 °C.
- Purify the PCR products using a solid-phase reversible immobilization (SPRI) bead isolation protocol: in new 1.5 mL tubes, add 56 µL of beads to each sample and proceed following manufacturer's instructions. Elute in 25 µL of Tris-HCl 10 mM. Libraries can be stored at -20 °C until use.
- Library check
- Use 1 µL of sample to assess library size using a bioanalyzer instrument.
- Use 1 µL of sample to quantify library molarity using a fluorescence-based Real time PCR assay, according to manufacturer's instructions.
- Library dilution and sequencing
- Dilute libraries to 10 nM using Tris-HCl 10 mM. For pooling libraries, transfer 5 µL of each diluted library to a new 1.5 mL tube and then dilute the pool to 4 nM in Tris-HCl 10 mM.
- Mix 5 µL of diluted pool with 5 µL of 0.2 N NaOH in a new 1.5 mL tube, vortex briefly, spin-down and incubate for 5 min at room temperature to denature the libraries.
- Put tubes on ice and add 990 µL of pre-chilled hybridization buffer (HT1). Aliquot 300 µL of denatured pool in a new 1.5 mL tube and add 300 µL of pre-chilled HT1 to obtain a 10 pM final library pool.
- In parallel, mix 2 µL of PhiX control library (10 nM) with 3 µL of Tris-HCl 10 mM in a 1.5 mL tube. Add 5 µL of NaOH 0.2 N, vortex briefly, spin-down and incubate for 5 min at room temperature to denature the diluted PhiX. Put tube on ice and add 990 µL of pre-chilled HT1.
- Aliquot 300 µL of denatured PhiX in a new 1.5 mL tube and add 300 µL of pre-chilled HT1 to obtain a 10 pM final PhiX.
- In a new 1.5 mL tube, mix 510 µL of denatured library pool with 90 µL of denatured PhiX library, thus obtaining a final 10 pM pool with 15% of PhiX control.
- Pipette these 600 µL of sample volume into the Load Sample reservoir of the thawed sequencing reagent cartridge and proceed immediately to perform a single-read 150-cycle run.
NOTE: The critical reagents and primer sequences required for this protocol are listed in Table 1.
Workflow of the retroviral scanning procedure
The workflow of retroviral scanning procedure is schematized in Figure 1. The target cell population is purified and transduced with a mLV-derived retroviral vector expressing an eGFP reporter gene. The transgene is flanked by the two identical long terminal repeats (5' and 3' LTR), ensuring synthesis, reverse transcription and integration of the viral genome into host DNA. The transduction efficiency is assessed by FACS analysis of eGFP expression. The cell population containing a high proportion (>30%) of mLV-transduced cells is amplified and subsequently lysed to extract genomic DNA containing the integrated mLV viral cassettes. Genomic DNA is digested and ligated with a compatible linker and the junctions between the viral 3' LTRs and the host genome are amplified by LM-PCR. Virus-host genome junctions are then massively sequenced using Roche or Illumina platforms. Finally, mLV integration sites are mapped to the human genome to define genomic clusters of recurrent insertion sites.
Amplification of mLV integration sites by LM-PCR
The LM-PCR is schematized in Figure 2. Genomic DNA is extracted from mLV-transduced cells and digested with the Tru9I restriction enzyme, which cuts frequently the human genome, generating fragments with a median length of 70 bp. A second restriction enzyme (PstI) is used to prevent amplification of integrated and non-integrated internal 5' LTR fragments.A Tru9I double-stranded linker is then ligated to the genomic fragments and LM-PCR is performed with primers specific for the linker and the 3' LTR to amplify the virus-host genome junctions. Nested PCR can be performed using primers compatible with Roche or Illumina sequencing platforms.
Analysis of virus-host genome junction amplicons
In the experiment represented in Figure 3, we purified CD34–CD13+ myeloid progenitor/precursors (MPP) and transduced them with an mLV-derived retroviral expressing the eGFP reporter gene. More than 60% of MPP cells expressed eGFP 48 h after transduction (data not shown). 15 days after transduction, we collected the cells, extracted gDNA and amplified the virus-host genome junctions, as described above. An aliquot of the pooled LM-PCR products was loaded on an 1% agarose gel to verify the presence and the size of the amplicons. We successfully visualized a DNA smear corresponding to the LM-PCR products of different sizes, ranging from 150 to 500 bp (Figure 3A). Amplicons were then concentrated by DNA precipitation and loaded on a 1% agarose gel. LM-PCR products were gel-purified and run on a bioanalyzer system, confirming the expected amplicon sizes (between 150 and 500 bp; Figure 3B).
Mapping of mLV integrations into active and cell-specific regulatory regions
In the experiment reported in Figure 4, hematopoietic stem/progenitor cells (HSPC), erythroid progenitor/precursors (EPP) and myeloid progenitor/precursors (MPP) were transduced with an mLV-derived retroviral vector. These results were obtained processing and sequencing samples derived from different cell types separately, to avoid the potential contamination/collisions18,19. Raw sequence reads generated by massive sequencing were processed by an automated bioinformatics pipeline to eliminate viral and linker sequences. Then, unique sequences of at least 20 bp were mapped on the human genome using Blat17. Raw alignments were filtered requiring the match to start within the first 3 nucleotides, univocal matches and a minimum of 95% identity. Clusters of recurrent mLV integrations were defined by a statistical comparison with a dataset of random genomic sequences, generated randomly extracting genomic positions from the human genome with a Tru91 restriction motif at a distance compatible with the sequencing platform. Control sequences were then processed through the same mapping and filtering pipeline used for integration sequences, to generate a random set of unique sites. To define mLV clusters, we applied the DBSCAN clustering algorithm19, comparing the distribution of consecutive mLV integrations with that of an equal number of random sites to identify regions of highly clustered integrations, which define cell-specific regulatory elements7,14,15. In order to avoid the generation of false clusters, multiple extractions from the random control dataset were performed. We mapped by LM-PCR and pyrosequencing 32,574, 27,546 and 36,358 mLV integration sites in HSPC, EPP and MPP, respectively. Clusters of recurrent mLV integrations co-mapped with acetylated enhancers and promoters (Figure 4A). Most of the mLV-targeted regulatory regions were cell-specific, such as: (i) the promoter of HSPC-specific SPINK2 gene (Figure 4B); (ii) the Locus Control Region containing potent enhancers of the erythroid-specific β-like globin genes (Figure 4C); (iii) enhancers located upstream of the MPP-specific LYZ gene (Figure 4D). Finally, we used luciferase assays to validate a subset of putative cell-specific mLV-targeted enhancers in EPP and MPP (Figure 4E).
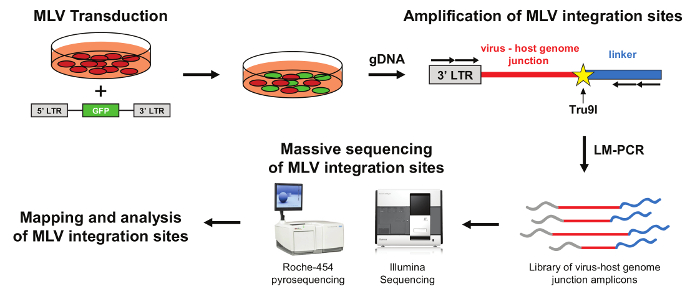
Figure 1: A general scheme of the retroviral integration site mapping procedure. Target cells are transduced with a mLV-based retroviral vector containing a eGFP cassette. Genomic DNA obtained from transduced cells is digested with Tru9I and ligated with a compatible Tru9I double-strand linker. mLV integration sites were amplified by nested LM-PCR and the library of virus-host genome junctions can be massively sequenced using Illumina or Roche platforms. The resulting reads were mapped to the human genome to define clusters of recurrent mLV integrations. Please click here to view a larger version of this figure.
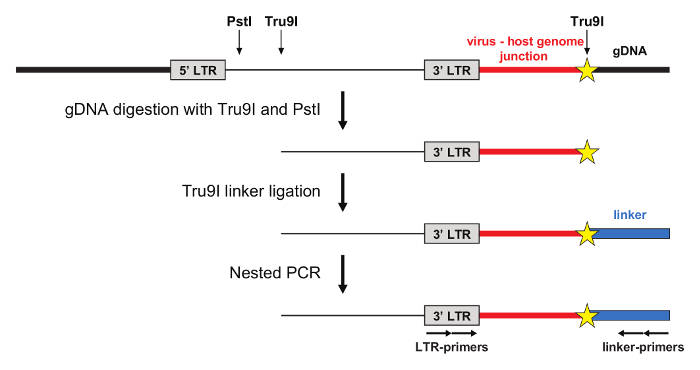
Figure 2: Amplification of virus-host genome junctions by LM-PCR. Genomic DNA (gDNA) containing the integrated mLV provirus is digested with Tru9I and PstI restriction enzymes, and ligated with a compatible Tru9I linker. Nested PCR is performed using primers specific for the LTR and the linker. Tru9I and PstI restriction sites in the viral and in the human genome are indicated. Please click here to view a larger version of this figure.
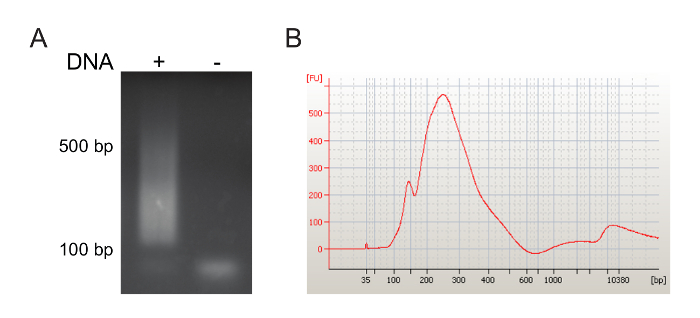
Figure 3: Analysis of LM-PCR amplicons. (A) LM-PCR products (lane +) were run on a 1% agarose gel and visualized by ethidium bromide staining. A no template sample served as negative control sample, where only PCR primers were visualized (lane -). (B) After gel purification, LM-PCR product size was checked by microcapillary electrophoresis. Sample size (bp) and fluorescence intensity (FU) are shown on x and y axes of the electropherogram, respectively. Please click here to view a larger version of this figure.
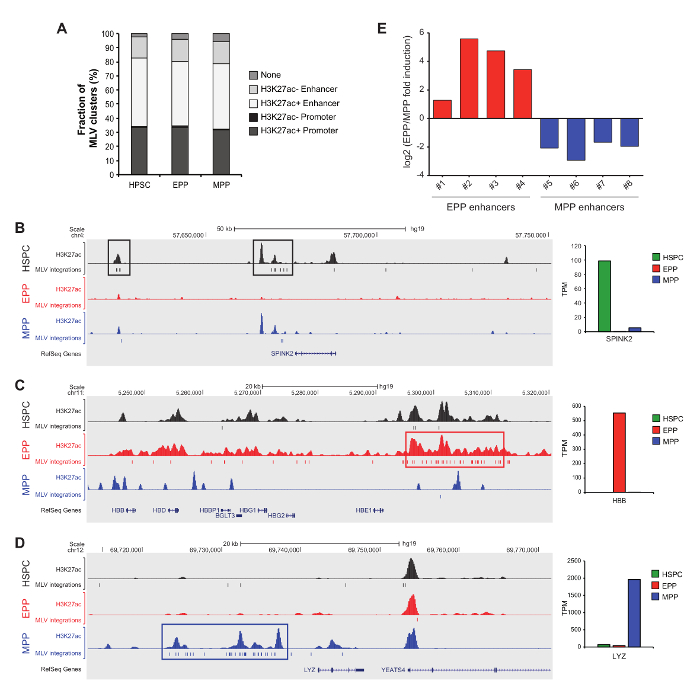
Figure 4: Mapping of mLV integration into epigenetically-defined regulatory regions. (A) We defined 3,498, 2,989 and 4,103 clusters of recurrent mLV integration sites in HSPC, EPP and MPP, respectively. In each cell population, >95% of mLV clusters overlapped with epigenetically defined enhancers and promoters. (B, C, and D) Cell-specific mLV-targeted regions were highly acetylated and associated with cell-specific expression of the targeted gene (SPINK2, HBB and LYZ, almost exclusively expressed in HSPC, EPP and MPP, respectively, as determined by Cap analysis of Gene Expression). mLV single integrations are depicted with small bars. TPM indicates Tag Per Million. (E) Putative cell-specific mLV-targeted enhancers in EPP and MPP. Figure 4 is adapted from reference14. We received the permission to re-use this figure under the creative commons license. Please click here to view a larger version of this figure.
