A Southern blot was done to verify that DARS2 was distributed randomly throughout the chromosome in the transposon library (t = 0) and that the fittest clones would persist over time. The Southern blot was performed on DNA extracted from the initial transposon pool (at t = 0) and every estimated 100 out of 700 generations of competition (Figure 3). Here, the total cellular DNA from each time-point was digested with the PvuI restriction enzyme, known to cut transposon NKBOR::DARS2 once only in a region not covered by the probe. The Southern blot was probed with a radioactively labeled DNA fragment complementary to part of NKBOR. As seen from Figure 3, the initial DARS2 pool (t = 0) lacked distinct bands, which shows that DARS2 was inserted randomly throughout the chromosome. Over time, a pattern emerged where the initial large pool of DARS2 clones developed into only one or a few persisting DARS2 clones (Figure 3; t = 0 to t = 700).
In the example shown, DARS2 insertion sites from the competition experiment were identified using WGS and easy gene walking. Here, WGS was used to identify insertion sites from the start pool (t = 0) and after 300, 400, and 700 generations of competition. Note that the coverage in the present deep sequencing was insufficient for a complete mapping of insertion sites at t = 0; however, it gives a representative subset of the total number of insertions. WGS confirmed the Southern blot result (i.e., the selection of the fittest DARS2 clones), ending with approximately 98% of all DARS2 insertions close to the wildtype DARS2 chromosomal location (DARS2 Clone IR and Clone rppH), while the remaining 2% were elsewhere on the chromosome (Figure 4). This strongly suggests that the wildtype position is optimal for DARS2 function. At t = 400, an insertion was found on the opposite replication arm with an almost identical distance to oriC as the wildtype DARS2 position (Figure 4), but this insertion was not recovered after 700 generations. Thus, replication-associated gene dosage cannot be the single determinant for optimal position.
Easy gene walking was used to identify DARS2 insertions sites in single clones isolated after 700 estimated generations of competition. Here, the two DARS2 insertion sites mentioned above (DARS2 Clone IR and Clone rppH) were identified. Easy gene walking was only done on 20 clones, and this explains why all DARS2 insertions sites mapped in WGS were not identified. DARS2-deficient cells were previously shown to initiate replication in asynchrony and to have a decrease in origin concentration relative to wildtype cells4,16,17,18. We therefore used flow cytometry to resolve the synchrony in the initiation of DNA replication and cellular origin content for the two selected strains (DARS2 Clone IR and Clone rppH) which, in both cases, were restored to wildtype levels (Figure 5). A representative example of a strain possessing a single copy of DARS2 located in the terminus is shown (Figure 5E). Here, the presence of a DARS2 element in the terminus does not restore synchrony or the cellular origin content to wildtype levels, while the selected DARS2 clones IR and rppH do.
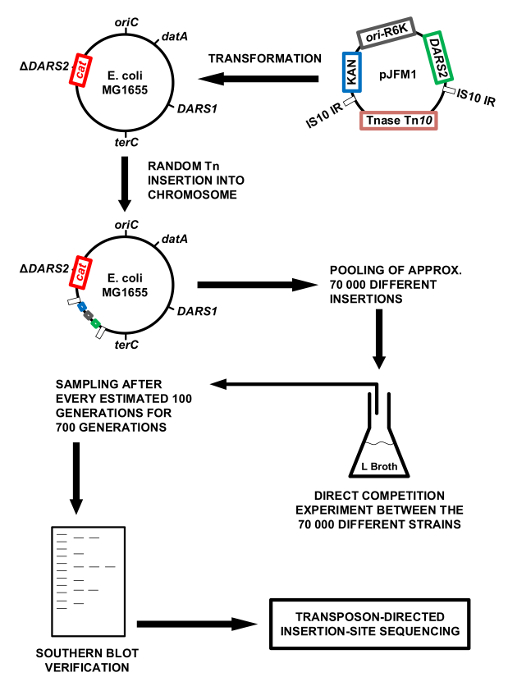
Figure 1: Methodology overview. Schematic presentation of the methodology. The chromosomal DARS2 region is cloned into the mini Tn10 on pNKBOR, creating pJFM1. pJFM1 is transformed into E. coli MG1655 ΔDARS2, which triggers a random insertion of DARS2 linked to Tn10 onto the chromosome of E. coli MG1655 ΔDARS2. Approximately 70,000 clones, each containing a different chromosomal DARS2 insertion, were pooled and competed directly against each other in LB broth at 37 °C. The direct competition experiment in LB broth was performed for an estimated 700 generations, where a sample was isolated for each 100 generations of direct competition. The total DNA was extracted from each isolated sample and used for Southern blotting and the identification of DARS2 insertions by WGS. Please click here to view a larger version of this figure.
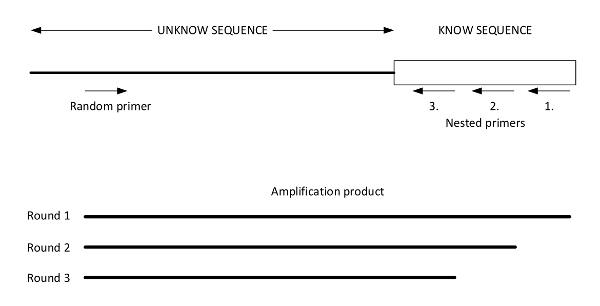
Figure 2: Graphic presentation of easy gene walking. This figure illustrates a genomic DNA template of an unknown DNA sequence adjacent to a known sequence with priming sites for the random primer and Nested Primers 1, 2, and 3. The results of the three successive amplifications performed using the three designed nested primers are illustrated below. The final product (from round 3) is sequenced using Nested Primer 3. This figure was adapted from Harrison et al.27. Please click here to view a larger version of this figure.
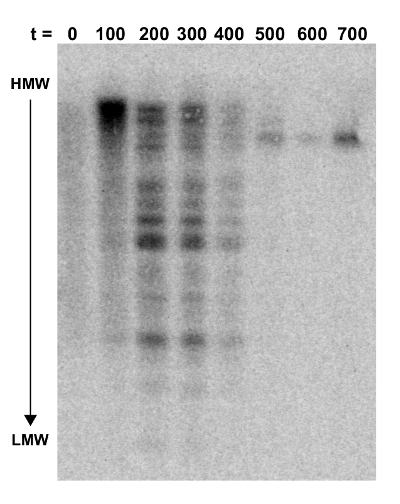
Figure 3: Southern blot probed for NKBOR. Southern blot analysis of DARS2 insertions into a DARS2-deficient strain. Genomic DNA extracted from every ~100 generations of direct competition, starting at t = 0 and ending at 700 generations, were digested with PvuI and gel-fractionated. The blot was hybridized with a NKBOR probe (see the Protocol). t indicates the number of generations of competition. This figure was adapted from Frimodt-Møller et al.4. HMW and LMW are high-molecular weight and low-molecular weight DNA, respectively. Please click here to view a larger version of this figure.
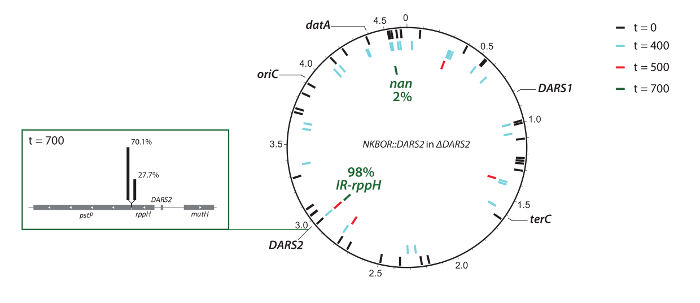
Figure 4: Graphic representation of resolved transposon insertions sites in ΔDARS2. The positions of oriC, datA, DARS1, DARS2, and terC are indicated. DARS2 insertion sites in ΔDARS2 at t = 0 (black bars), t = 400 (light blue bars), t = 500 (red bars), and t = 700 (green bars), resolved by full-genome sequencing. This figure was made using DNAPlotter33 and was adapted from Frimodt-Møller et al.4. Please click here to view a larger version of this figure.
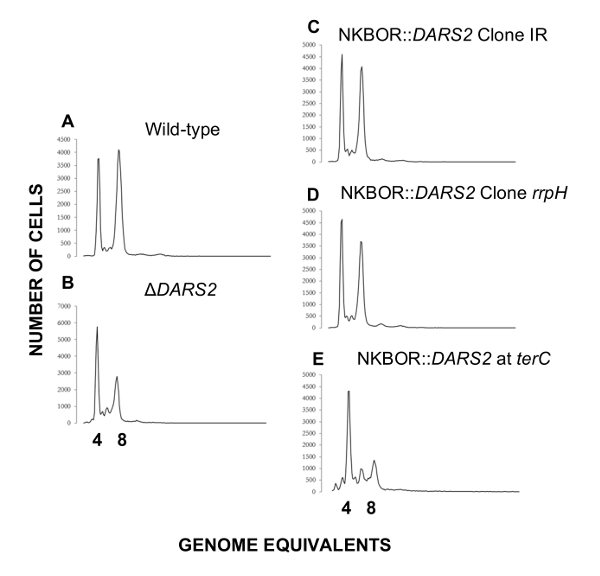
Figure 5: Representative flow cytometry histograms of transposon sites found by easy gene walking at t = 700. Cells were grown in AB minimal medium supplemented with 0.2% glucose, 10 µg/mL thiamine, and 0.5% casamino acids at 37 °C. Wildtype and ΔDARS2 are shown in A and B, respectively. Derivatives of the wildtype strain MG1655 devoid of DARS2 at the original locus and instead carrying a copy of DARS2 at the resolved transposon site rrpH, IR, and the terminus (terC) are shown in C, D, and E, respectively. This figure was adapted from Frimodt-Møller et al.4 to show a DARS2 location that results in a cell cycle anomaly (terC) or that restores the wildtype phenotype(rrpH, IR). Please click here to view a larger version of this figure.
