To validate the application of 3D small intestinal mouse organoids as a model to quantify the effect of compounds regulating the intestinal barrier integrity, we applied IFN-γ. To do so, we isolated and cultured organoids derived from IFN-γ responsive wild type and IFN-γ-receptor-2 knockout mice, which do not respond to IFN-γ8. Upon treatment for 48 h with IFN-γ or PBS (control), all organoids were exposed to LY and imaged by confocal spinning disc live cell microscopy in 5 min intervals for a period of 70 min. The functional integrity of the intestinal barrier in this model resulted in exclusion of LY from the organoid's lumen while intraluminal accumulation of LY signified destruction of the TJ. The representative fluorescence microscopic images after 70 min of incubation with LY clearly demonstrate that intraluminal LY fluorescence was only visible in organoids from wild type animals treated with IFN-γ. In unstimulated (PBS) controls nor in organoids derived from knock out animals (IFN-γR2ΔIEC, Figure 1), no intraluminal LY fluorescence was present after 70 min.
The addition of EGTA causes an unspecific breakdown of the intestinal barrier integrity by sequestering TJ cofactors. This control was always utilized at the end of the experiment to demonstrate the ability of the respective organoid to take up LY (Figure 1). If no intraluminal LY fluorescence was detected upon EGTA treatment, the organoid was excluded from the experiment.
For the quantitative evaluation of the microscopic results, LY fluorescence was measured within the organoid's lumen and outside of the organoid. Relative intensity values were calculated (fluorescence inside/ fluorescence outside + inside) and are shown for each time point imaged. It is recommended to avoid imaging of organoids of varying sizes. We chose to focus on organoids with a diameter of 80 ± 30 µm (Figure 2). A schematic of the protocol with representative images is shown in Figure 3. Some major problems and troubleshooting techniques are shown and discussed in Figure 4.
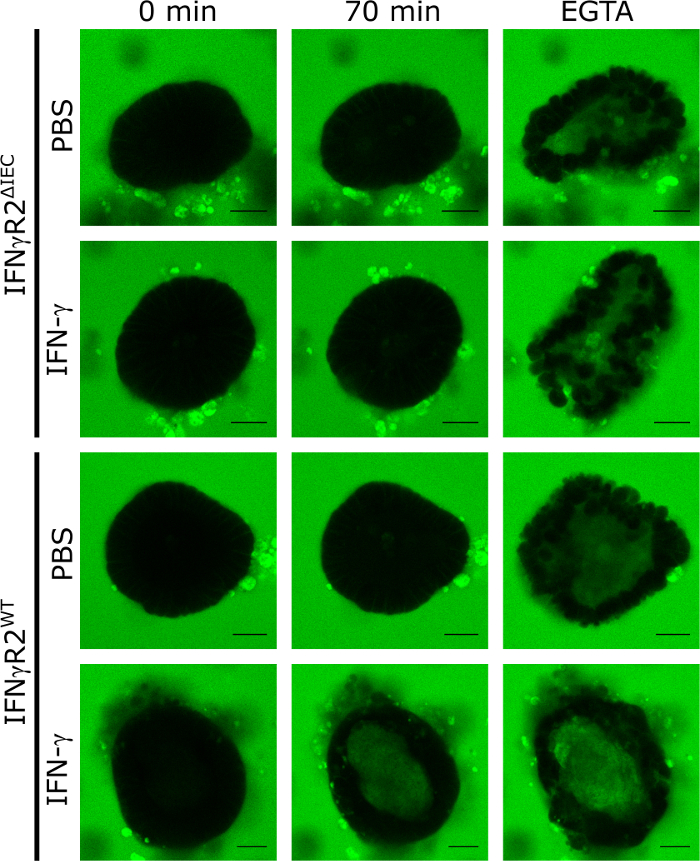
Figure 1: Intestinal barrier integrity can be analyzed in mouse organoids. Intestinal organoids from IFN-γR2WT and IFN-γR2ΔIEC were cultured in the presence of IFN-γ for 48 h or left untreated. To investigate the integrity of the intestinal barrier, LY (457 Da) was added and confocal fluorescent images were captured in 5 min intervals for a total of 70 min. Representative images at time point 0 min, 70 min, and after addition of EGTA are shown (green = Lucifer yellow; Scale bar = 20 µm). This figure has been modified from Bardenbacher et al.8. Please click here to view a larger version of this figure.
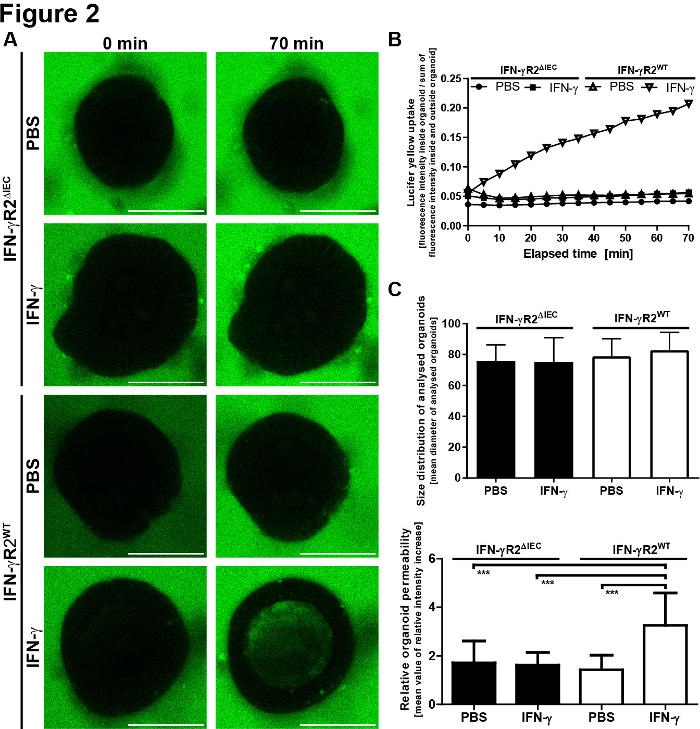
Figure 2: Small intestinal organoid barrier integrity model provides quantitative results. (A) LY fluorescence was determined inside and outside the organoid. Relative intensity values were calculated (inside/fluorescence outside + inside) relative to the initial relative intensity + SEM and are shown for each time point. (B) Size distribution of analyzed organoids. To reduce the standard deviation and errors due to changes in the surface-to-volume ratio, we only analyzed organoids with a diameter of 80 ± 30 µm. Mean values of the respective organoid diameters are shown + SD (IFN-γR2WT, n = 20; IFN-γR2ΔIEC, n = 18). The mean diameter values did not vary significantly between the different groups (one-way ANOVA). (C) The permeability of the organoids was determined 70 min after the addition of LY. It was defined by dividing the intraluminal fluorescence intensities after 70 min by the minimal relative fluorescence intensities measured during the observation period. Each bar represents mean values + SD, measured in 10 organoids derived from two independent experiments (IFN-γR2WT, n = 20; IFN-γR2ΔIEC, n = 18). IFN-γ significantly increased the LY uptake only in IFN-γR2WT organoids. ***p-value <0.001 in the Student’s t-test. This figure has been modified from Bardenbacher et al.8. Please click here to view a larger version of this figure.
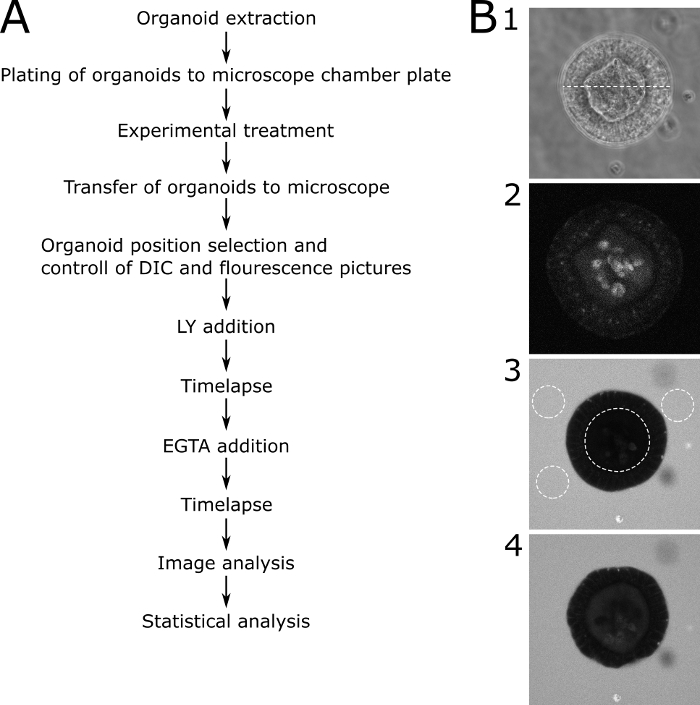
Figure 3: Schematic protocol with representative images. (A) Schematic description of the main steps of the protocol. (B) Representative pictures of the major steps of the protocol. (B1) DIC microscopy image of a central slice through a suitable organoid that was selected for permeability analysis. The dotted line represents a width of 89 µm. (B2) Fluorescence microscopy picture of the same organoid in (B1) before adding LY. The image shows the autofluorescence of the organoid. (B3) An organoid 70 min after the addition of LY. The depicted organoid shows no uptake of LY and therefore an intact barrier function. Dotted lines show the ROIs for further analysis. The inner lumen of the organoid and three representative areas around the organoid are marked. (B4) An organoid after the addition of EGTA. The organoid is usable for further analysis because it shows LY uptake after the EGTA treatment. Please click here to view a larger version of this figure.
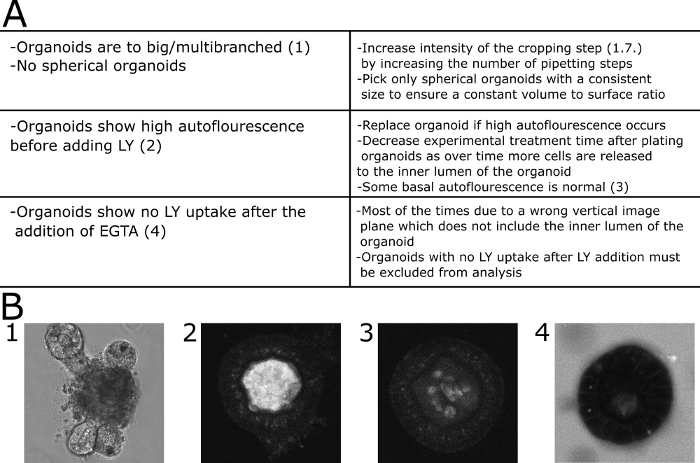
Figure 4: Troubleshooting of common problems. (A) Table with common problems and solutions. (B) Exemplary images. (B1) DIC image of a large multibranched organoid that is not suitable for this assay. (B2). Confocal image of an organoid displaying high autofluorescence before LY was added to the medium. The organoid was excluded from quantification. (B3) Confocal image of an organoid displaying low autofluorescence before LY was added to the medium. The fluorescence was quantified in this case. (B4) Organoid showing no LY uptake from the medium 30 min after addition of EGTA and therefore excluded from quantification. Please click here to view a larger version of this figure.
