Analysis of SEC-SAXS data via EFA deconvolution and Scatter
Summary
SEC-BioSAXS measurements of biological macromolecules are a standard approach for determining solution structure of macromolecules and their complexes. Here, we analyze SEC-BioSAXS data from two types of commonly encountered SEC traces—chromatograms with fully resolved and partially resolved peaks. We demonstrate the analysis and deconvolution using scatter and BioXTAS RAW.
Abstract
BioSAXS is a popular technique used in molecular and structural biology to determine the solution structure, particle size and shape, surface-to-volume ratio and conformational changes of macromolecules and macromolecular complexes. A high quality SAXS dataset for structural modeling must be from monodisperse, homogeneous samples and this is often only reached by a combination of inline chromatography and immediate SAXS measurement. Most commonly, size-exclusion chromatography is used to separate samples and exclude contaminants and aggregations from the particle of interest allowing SAXS measurements to be made from a well-resolved chromatographic peak of a single protein species. Still, in some cases, even inline purification is not a guarantee of monodisperse samples, either because multiple components are too close to each other in size or changes in shape induced through binding alter perceived elution time. In these cases, it may be possible to deconvolute the SAXS data of a mixture to obtain the idealized SAXS curves of individual components. Here, we show how this is achieved and the practical analysis of SEC-SAXS data is performed on ideal and difficult samples. Specifically, we show the SEC-SAXS analysis of the vaccinia E9 DNA polymerase exonuclease minus mutant.
Introduction
Biological macromolecules are too small to be seen even with the best light microscopes. Current methods to determine their structures generally involve crystallizing the protein or measurements on vast numbers of identical molecules at the same time. While crystallography provides information on the atomic level, it represents an artificial sample environment, given that most macromolecules are not presented in a crystalline form in the cell. During the last couple of years cryo-electron microscopy delivered similar high-resolution structures of large macromolecules / macromolecular complexes, but although the samples are closer to physiological condition, they are still frozen, hence immobile and static. Bio-small angle X-ray scattering (BioSAXS) provides a structural measurement of the macromolecule, in conditions that are relevant to biology. This state can be visualized as a low resolution 3-D shape determined on nanometer scale and captures the entire conformational space of the macromolecule in solution. BioSAXS experiments efficiently assess oligomeric state, domain and complex arrangements as well as flexibility between domains1,2,3. The method is accurate, mostly non-destructive and usually requires only a minimum of sample preparation and time. However, for the best interpretation of the data, the samples need to be monodisperse. This is challenging; biological molecules are often susceptible to contaminations, poor purification and aggregation, for example from freeze thawing4. The development of inline chromatography followed by immediate SAXS measurement helps mitigate these effects. Size-exclusion chromatography separates the samples by size thus excluding most contaminants and aggregations5,6,7,8,9,10. However, in some cases even SEC-SAXS is not sufficient to produce a monodisperse sample, because the mixture may consist of components that are too close in size or their physical properties or their fast dynamics lead to overlapping peaks in the SEC UV trace. In these cases, a software-based deconvolution step of the obtained SAXS data might lead to an idealized SAXS curve of the individual component5,11,12. As an example, in protocol section 2, we show the standard SEC-SAXS analysis of the vaccinia E9 DNA polymerase exonuclease minus mutant (E9 exominus) in complex with DNA. Vaccinia represents the model organism of the Poxviridae, a family containing several pathogens, for example the human smallpox virus. The polymerase was shown to bind tightly to DNA in biochemical approaches, with the structure of the complex recently solved by X-ray crystallography13.
Most synchrotron facilities will provide an automated data processing pipeline that will perform data normalization and integration producing a set of unsubtracted frames. But the approach described in this manuscript could also be use with a lab source provided SEC-SAXS is performed. Furthermore, additional automation may be available that will reject radiation-damaged frames and perform the buffer subtraction14. We will show how to perform primary data analysis on pre-processed data and make the most of the available data in section 2.
In section 3, we show how to deconvolute SEC-SAXS data and analyze the curves efficiently. While there are several deconvolution methods such as the Gaussian peak deconvolution, implemented in US-SOMO15 and the Guinier optimized maximum likelihood method, implemented in the DELA software16, these generally require a model for the peak shape12. The finite size of individual peaks we are investigating allows the use of evolving factor analysis (EFA), as an enhanced form of singular value decomposition (SVD) to deconvolute overlapping peaks, without relying on the peak shape or scattering profile5,11. A SAXS-specific implementation can be found in BioXTAS RAW17. EFA was first used on chromatography data when 2D diode array data allowed matrices to be formed from absorbance against retention time and wavelength data18. Where EFA excels is that it focuses on the evolving character of singular values, how they change with the appearance of new components, with the caveat that there is an inherent order in the acquisition10. Fortunately, SEC-SAXS data provides all the necessary ordered acquisition data in organized 2D data arrays, lending itself nicely to the EFA technique.
In section 4, we will demonstrate the basics of model-independent SAXS analysis from the buffer-background subtracted SAXS curve. Model-independent analysis determines the particle’s radius-of-gyration (Rg), volume-of-correlation (Vc), Porod Volume (Vp), and Porod-Debye Exponent (PE). The analysis provides a semi-quantitative assessment of the particle’s thermodynamic state in terms of compactness or flexibility via the dimensionless Kratky plot2,4,19.
Finally, SAXS data are measured in reciprocal space units and we will show how to transform the SAXS data to real-space to recover the pair-distance, P(r), distribution function. The P(r)-distribution is the set of all distances found within the particle and includes the particle’s maximum dimension, dmax. Since this is a thermodynamic measurement, the P(r)-distribution represents the physical space occupied by the particles’ conformational space. Proper analysis of a SAXS dataset can provide solution-state insights that complement high-resolution information from crystallography and cryo-EM.
Protocol
1. Protein expression, purification and SEC-SAXS measurement is based on the published protocol13
- Follow the inline SEC-SAXS data collection protocol (Brennich et al.6) in brief.
- Equilibrate the SEC-column with at least 2 column volumes of SEC running buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl).
- Prepare 50 µL of sample of E9 exominus at 8‒10 mg/mL with 20% molar excess of a partial dsDNA (TCAGGAAGATAACAGCGGTTTAGCC and GGCTAAACCGCTGTTATCTT). E9 exominus binds with an KD of 12 ± 6 nM (see Supplementary Data).
- Inject 50 μL of this mix onto a SEC-column (S200 Increase) inline with the flow cell for SAXS measurements at 0.3 mL/min.
- Collect 1000 frames at 1 s exposure each.
NOTE: On BioSAXS beamline BM29, at the European Synchrotron Facility (ESRF) the individual frames are processed automatically and independently within the EDNA framework14. After the data collection, open the ISPyB database20 and under the Data acquisition tab press the Go button to access the data set and the results of the automatic analysis21.
- Download the data.
2. Primary data analysis
- Open the Java-based program Scatter IV (see the Table of Materials) and perform a background subtraction for size exclusion chromatography (SEC) data.
- Open the SEC tab. Drag and drop the reduced data files (*.dat) into the “Drop Data below” window. Set output directory “Out Dir ::” by clicking the blue-labeled Output Dir button.
NOTE: If your data was collected in nm-1, a conversion box will need to be checked (bottom left of panel) when dropping files into the window or the subtraction tab. - Edit the experimental details, use the Edit Details button and fill out as many fields as possible, these include sections on which source/beamline was used to collect data, the collection parameters and sample details. These will be saved with the data and allow to more easily populate the “Data collection parameters” section in future publications.
- Enter the sample name in the Save as box. Click on TRACE.
NOTE: This has two effects. Firstly, it will create a *.sec file for the data. This is a single text file that will collate all the experimental observations from the separate *.dat files. In addition, the *.sec file contains the averaged set of frames that is the buffer-background, all the frames used in the averaging as well as the buffer-background subtracted frames across the entire SEC-SAXS experiment. Secondly, a signal plot is created which plots the frame number versus Integral ratio to the background. This shows the selected frames (gray) that were averaged for the buffer subtraction. The points for the average buffer are determined from the whole range of the data. However, it is advisable to manually choose the buffer frames for averaging as a poorly defined background may occur due to poorly equilibrated or dirty columns or capillary fouling22. - Select buffer frames manually. Click Clear Buffers then reselect a buffer region with a left click-drag, on the trace curve. Ideally, this should be a flat region before the void volume of the SEC column of approximately 100 frames. Click SET BUFFER and then Update to recalculate the *.sec file which may take a few minutes.
- Identify a region of interest (ROI). On the signal plot, select the region of the peak of interest, with a left click-drag.
NOTE: This populates three plots in the right-hand panel. The top two plots are linked with crosshairs moving between them, a second signal plot (top right) shows only the ROI selected, with the intensity of each frame in blue and the corresponding Rg of each frame in red and a corresponding heat map below, showing the residuals for each frame colored according to the Durbin-Watson auto-correlation analysis. Regions of high similarity are colored cyan (Durbin-Watson, d = 2) while dissimilar frames will follow darker blues to pinks and finally to reds depending on the severity of the dissimilarity (d > 2). The bottom plot is a subtracted I versus q curve for the central selected frame (also denoted by a vertical line). The arrow keys can be used to navigate through the subtracted frames. The I versus q plot will demonstrate the quality of the subtracted frames from the SEC experiment. - Select frames to merge. Click on the crosshairs in the heat map plot to select the subset of frames that will be used for merging. The crosshairs will identify a triangular area of predominately cyan that falls to the bottom right-hand side of the crosshairs. Use a mouse-click to set these frames as selected and highlight the frames in the corresponding area of the Signal plot above. These frames should ideally highlight a region with a stable Rg.
NOTE: As needed, zoom in on the heat map with a left-click drag and zoom out with a left-click swipe to the right. - When satisfied with the selected frames click MERGE. This will merge the subtracted frames and present them in the ANALYSIS tab.
- Open the SEC tab. Drag and drop the reduced data files (*.dat) into the “Drop Data below” window. Set output directory “Out Dir ::” by clicking the blue-labeled Output Dir button.
3. Data deconvolution
- Open the deconvolution program (e.g., BioXTAS Raw 2.0.0).
- In the deconvolution program, load the dataset, under Files tab, in the Control Panel, use foldether symbol to locate the data or copy and paste the location into the address bar.
NOTE: Make sure the folder contains only the raw *.dat files and no processed or average data files. - Highlight all the *.dat files, hit the Plot Series button, a plot of integrated intensity versus frame number will be drawn in the “Series Plot”.
- In the Control Panel select the Series tab and then click to highlight the curve. Open up the LC Analysis pop-up window using the button at the base of the control panel. This window gives access to several options, such as selecting varying molecule types (protein or RNA). It also allows the user to select the buffer region for the plot. In the first instance click Auto; this should select a suitable buffer region.
NOTE: If this fails, possibly due to an unstable baseline, then “Add region” to optimize the buffer region. This populates the Buffer box with a smaller box in which one can manually add the frame numbers to use for the buffer. Alternatively, click “Pick” to give the option of selecting an area on the plot. Locate the area, left-click once for the start position, move the cursor to next position and left-click again. It may be necessary to add more than one buffer location. Click Set buffer and the curves will be subtracted and the Rg calculated across the SEC peak. If a pop-up box appears, click OK. - To start the Evolving Factor Analysis (EFA), right-click on the highlighted file at the bottom of the Control Panel and then select EFA from the menu.
- Check that a pop-up window opens which shows the single value decomposition (SVD) of the data set. In the controls box, check the Use Frames box so that the whole peak area to deconvolute is covered in the intensity plot. The “Singular Values” plot, top right, shows the intensity of the singular values (separate peaks/species) above the baseline.
NOTE: The number of points present above the baseline represents the number of scattering species present. With the caveat that it is the relative magnitude of the singular value to the flat area/baseline that matters. - To help validate the number of single values, use the bottom AutoCorrelation plot. This shows the right and left single correlation vectors. Click Next.
NOTE: These essentially represent scattering or concentration profiles for the vector in the solution. Where the absolute size represents the significance of the vector. A significant component will have an autocorrelation near 1 (a rule of thumb cutoff is >0.6‒0.7). RAW helpfully calculates this and is shown in the #Significant SVs box, bottom left, though you can change this if necessary. If there are several single values (e.g. 4+), it may be necessary to look at just 2 or 3 of the components only, altering the range of the data used. The lower the number of components the easier the EFA analysis will be but at the cost of using less data. In complex situation, where the left and right singular vectors, which should be similar, do not match reduce the significant SV number and decrease the number of frames used until the left and right singular vectors are similar. - Check that the EFA is calculated by generating plots in the forward and backward directions for each vector. These plots show when components start (forward plot) and exit (backwards plot) the solution profile for the selected SEC-SAXS data. RAW tries to identify these ranges; change these using the arrows next to the counters so that each circle is at the start of an inflection point rising from or falling to baseline. Click Next.
NOTE: The last stage of the EFA turns the SVD vectors back into scattering curves. On the left of the window, the previously defined ranges are plotted at the top. These ranges are the constraints to define where to rotate the singular vectors back into scattering curves. The right hand panel shows these corresponding scattering curve profiles, for each separated peak. A plot for the concentration of each peak, that should be representative of elution profiles and a plot for the mean error weighted chi2. The chi2 plot is measuring the deconvolution data set to the original data set. Ideally, this will be flat, however spikes can often be seen. - Try to reduce or eliminate spikes by altering the Component Range Controls, first identify approximately which frame corresponds to the spike (from chi2 plot) and then, in the Range Controls, which component contains this frame (it could be more than one), using the arrows, move up or down the corresponding range.
NOTE: This should produce a response, increasing or decreasing the spike. If the spike frame was present in more than one component then a little trial and error between each component may be necessary. - When a minimum chi2 has been achieved, perform a validation check by clicking back, the previous window appears to allow verifying if the changes made have drastically changed the original EFA plots. If they still look valid, click Next. Click Save EFA Data to save the plots and then click Done; to close the EFA window.
NOTE: A second validation is to click off the check-box next to each component range, in turn. These provide a positive concentration constraint to each component and turning off will check if these significantly affect the data set. If no change is seen in the concentration plot then the data are valid.
- Check that a pop-up window opens which shows the single value decomposition (SVD) of the data set. In the controls box, check the Use Frames box so that the whole peak area to deconvolute is covered in the intensity plot. The “Singular Values” plot, top right, shows the intensity of the singular values (separate peaks/species) above the baseline.
- Back in the RAW window, click the Profiles tab in the Control Panel to view the curves and in the Manipulation tab of the Control Panel, manipulate the curves further or save the curves as *.dat files by right-clicking on the file and selecting save selected file(s) from the menu pop up. Save the file. Use Scatter IV for further analysis.
NOTE: Further information and instructions on deconvolution and EFA BioXTAS RAW is found at https://bioxtas-raw.readthedocs.io/en/latest/
4. Determine SAXS properties
NOTE: An in-depth tutorial for SAXS determination is found at Bioisis.net. Here we show a basic step by step approach, highlighting the most useful buttons in Scatter.
- In the Scatter ANALYSIS tab, press the G button for the manual Guinier analysis tool, to the right of each sample file. The plot that opens shows the ln[I(q)] versus q2 in the top box and the corresponding residuals in the bottom box. Add or remove points such that the residuals do not have a “smile” or “frown” feature. The selected data in the Guinier fit should not exceed the maximum q x Rg limit of 1.3.
- Press the Normalized Kratky button; the plot that pops up provides a semi-quantitative assessment of macromolecule’s structural state, normalized for mass and concentration.
NOTE: The crosshairs designate the Guinier-Kratky point at (√3, 1.1)19. A compact, spherical protein will show a single peak with the maximum value at the Guinier-Kratky point. An intrinsically disordered or cylindrical biopolymer would have a maximum greater than the crosshairs and would not decrease. A protein that had both folded domains and long elongated unstructured regions might present with an increased maximum through the crosshairs but would also show an obvious decreasing trend at higher q x Rg. - Click on the Vc button (Volume-of-correlation), which brings up two plots, the total scattered intensity and an integrated area of the total scattered intensity as a function of q. The plots are used as a quick reference to validate the quality of the scattering curve.
NOTE: The total scattered intensity is sensitive to the I(0) and if this has not been measured correctly then the plot will not show a continuous line. The integrated area plot, ideally, should show a sigmoidal line with an extended plateau for each SAXS curve. If there are buffer mismatch/subtraction, aggregation or interparticle interference in the sample a sharp slope will be observed at higher q-values. - Press the Flexibility button to start the flexibility analysis. This will open a window with four panels and a slider at the bottom. Each opened panel shows a plot exploiting a power-law relationship that exists between compact and elongated/flexible biopolymers23. To use, move the slider at the bottom of the box from right to left with the left mouse button pressed. Keep moving slowly to the left until a plateau in one of the plots is reached.
NOTE: If the plateau is seen in the Porod-Debye plot, then the sample is compact in nature, which should be consistent with a single peak at the Guinier-Kratky point in a normalized Kratky plot. If the plateau is reached first in the Kratky-Debye plot then the sample is most likely elongated or flexible. If the SIBYLS plot is first to plateau, then the sample most likely contains areas of both compactness and flexibility, a particle with mixed states. The theory for this flexibility relationship with the Porod-Debye Law is exquisitely addressed in Rambo, et al.23 - Click on Volume. Volume determination should be performed immediately after the flexibility analysis from above. When opened after the flexibility analysis, a pop up with three more graphs is generated. In the bottom, left corner the Porod-Debye plot remembers where one left the slider from the flexibility plot, showing the plateaued area.
- To calculate the volume of the particle, move the start and endpoints using the arrow buttons or type in the boxes, so that the blue line on the plot fits the plateaued region. For an unbiased result, the residuals in the top right Porod-Debye exponent power-law fit, should show no pattern.
- Press the P(r) tab. The real-space distribution is in the left panel and the scattering curve for the sample in the right-hand panel. The objective is to create a real-space representation of the sample from the reciprocal space SAXS curve. Ideally, the distribution curve will be smooth with no waves present and should just gently kiss the x-axis.
NOTE: The instrument’s measured q-range may not be entirely useable due to poor buffer matching, aggregation, radiation damage, sub-optimal exposure times and low particle concentrations. The P(r)-determination step will fundamentally determine the useable qmin and qmax range of the SAXS dataset and it should be this range of data that is used for any subsequent modeling or fitting.- Right-click on the sample name then click Find DMAX to open a new window. Limits for the dmax are pre-set with the suggested qmax (maximal data points used), lower and upper dmax limits and a lower and upper alpha score. Three models can be chosen (L1-norm, Legendre and Moore) and the use of background included. Leave these unchanged in the first instance.
- Press the Start button. A composite distribution is created in the left panel with the suggested dmax and alpha level written underneath. If this looks acceptable then close the window and return to the P(r) tab. The reciprocal space plot will have been cropped to match the suggested qmax.
- Choose the model Moore, click on Background and then set the alpha level and dmax to the suggested values from the pop-up box. Press the refine button. A cross-validation plot will pop up showing if any points had to be rejected, marked in red. If there are only a few points rejected and the distribution looks good then the model is good.
NOTE: The cross-validation plot will highlight regions of the data that are inconsistent with the determined P(r)-distribution. If the rejected region is mainly in the low-q region, that is the region near the y-axis, this likely suggests a dmax that is too short, presence of aggregation or higher order oligomers. It highlights an inconsistency between the higher and lower resolution information. Here, dmax and qmin (increasing start value) should be adjusted using a manual, trial-and-error approach. Similarly, if the rejected region is mainly in the high-q region, this may indicate an issue with the background subtraction or that the signal is too weak to be meaningfully explained by the determined P(r)-distribution. In this case, qmax should be truncated (decreasing end) until no additional data are rejected. Ideally, rejected points should be randomly distributed and make up less than 5% of the useable data. A properly defined qmin, qmax and “dmax” will produce a smooth distribution where the dmax kisses the x-axis. However, do not increase this value so much that it completely removes the Guinier region. This point is easily found by checking the q x l(q) box (on the left of the panel above the table). The scattering curve is replaced by the “Total Scattered Intensity plot”, on this curve all points before the max inflection are part of the Guinier region. After removing points try again to increase/decrease the “dmax” and then refine once more. If problems persist, especially when many points are being rejected from the start of the validation curve, this strongly suggests the data are not ideal for structural modeling.
- To print a report, navigate back to the Analysis tab, left-click to highlight the sample then right-click on the sample name and move to Create report from single data set in the menu. A text box opens to allow for comments to be added. A PDF document is produced showing all the figures and values generated.
Representative Results
The advantage of using deconvolution over classical frame selection13 is to remove the influence of species on one another, producing a monodisperse scattering signal. This is also often followed with a better signal to noise ratio. When E9 exominus is bound to DNA and run using SEC-SAXS, two peaks are observed (Figure 1). The first, large peak (approximately frames 420‒475) is the E9 exominus-DNA complex the second (approximately frames 475‒540), the unbound state (see Supplementary Data: Figure 2). While the classical approach of selecting frames provides a stable Rg of the complex in the first peak (see Supplementary Data: Figure 3), the second peak is clearly merged and the Rg across the plot shows that the second peak of interest does not have a stable Rg, due to cross-peak contamination. Only 5 frames could be used that showed a semi-stable Rg, when subtracted they gave an Rg = 36.3 Å (Figure 2, green). When the peaks were deconvoluted using EFA the corresponding curve for the second peak (Figure 2, blue) was overlaid with the original and showed a clear decrease in signal to noise, and a lower Rg, 34.1 Å was recorded. The Kratky plot (Figure 3) shows the complex with the deconvoluted peak (blue) is more globular. This is confirmed by the P(r) curve (Figure 4) which gives a dmax 108.5 Å for the deconvoluted curve (blue) while the non-deconvoluted is more elongated with a dmax 120 Å (green), this is most likely due to heterogeneity arising from the unbound E9 exominus.
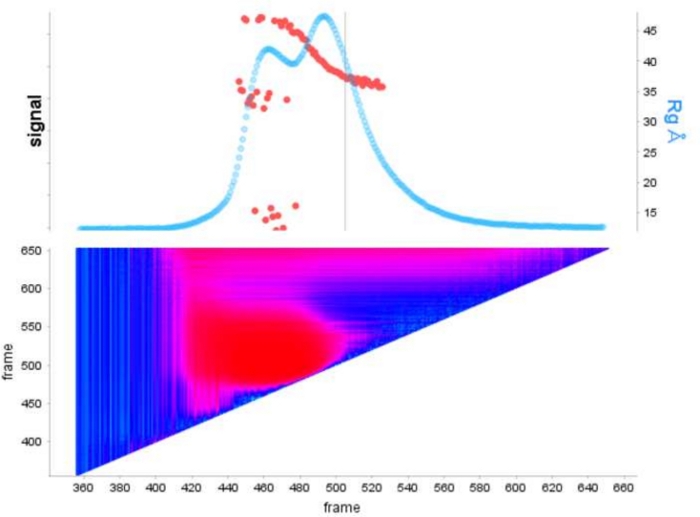
Figure 1: Signal plot of E9 exominus alone and with DNA in complex.
The top panel shows a plot of the integral ratio to the background for each frame of a SEC-SAXS run (light blue). The red points show the Rg at each frame over the peak. The bottom panel shows the corresponding heat map showing the residuals for each frame colored according to the Durbin-Watson auto-correlation analysis, regions of high similarity are colored cyan while dissimilar frames follow darker blues to pinks and finally to red depending on the severity of the dissimilarity. Please click here to view a larger version of this figure.
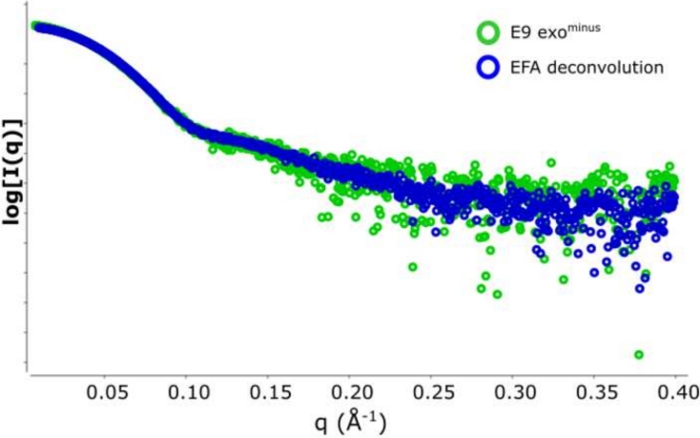
Figure 2: Plot of intensity versus scattering vector.
An overlay of the subtracted SAXS data form the E9 exominus . In green 5 frames (frame 517‒522) averaged and subtracted from an area of semi-stable Rg and in blue the representative scattering curve derived from the EFA deconvolution of the SEC-SAXS peak. Please click here to view a larger version of this figure.
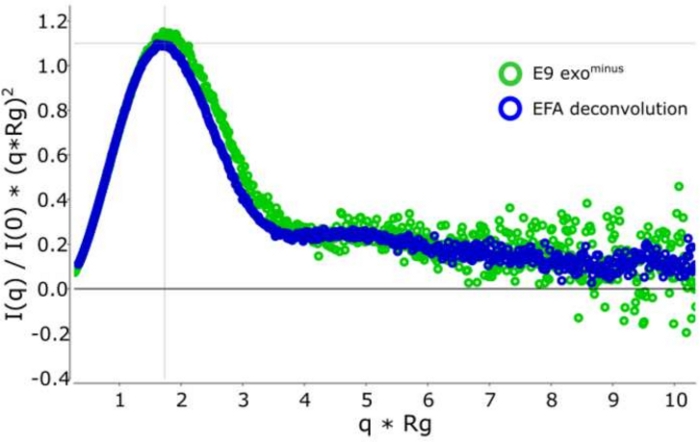
Figure 3: Dimensionless Kratky curve.
Overlay of the deconvoluted (blue) and non-deconvoluted (green) Kratky curve showing E9 exominus is globular. Please click here to view a larger version of this figure.
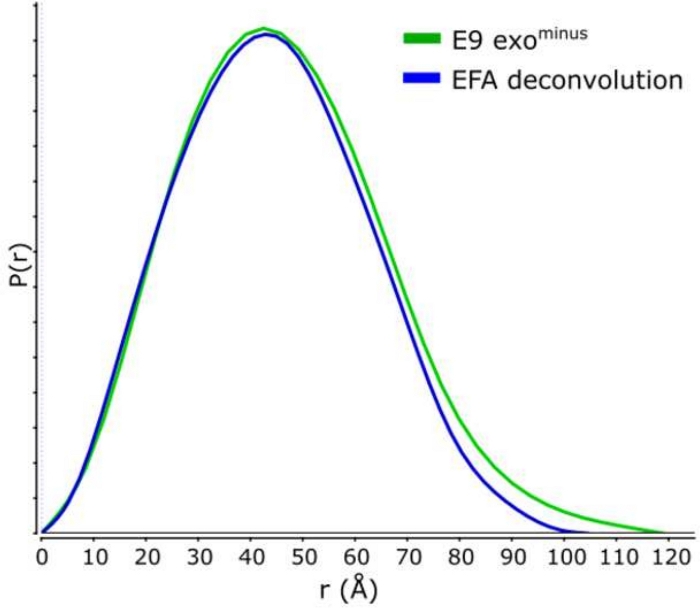
Figure 4: P(r) curve.
The overlay of the deconvoluted (blue) and non-deconvoluted (green) curves for the E9 exominus. Please click here to view a larger version of this figure.
Supplementary Data. Please click here to download this file
Discussion
It is desired to have a monodisperse sample before starting a SAXS experiment, but in reality, many data collections do not satisfy this and must be improved by combining the measurement with inline chromatography—SEC in most cases. However, even the shortage of time between purification and data acquisition monodispersity of the sample is not guaranteed. Most commonly, this applies to experiments where components are too close in size or in their physical properties to be separated or are prone to fast dynamics. Here, we have provided a protocol combining single value decomposition with evolving factor analysis to remove the influence of DNAbound E9 exominus from its unboundform creating a monodisperse scattering profile that we were then able to analyze with the SAXS package Scatter IV.
SVD with EFA of SEC-SAXS data are very powerful methods developed to deconvolute SAXS data and improve analysis, but they do have limitations. They require that noise or drift in the buffer baseline of the SEC-SAXS is kept to a minimum. This may involve extra column equilibration (better to use more than 3 column volumes, depending on the buffer) before sample loading. However, the most critical step is the choice of the number of the singular values and the range of data used, as this will greatly affect the accuracy of the deconvolution. It is for this reason that the results should not be taken on their own but further analyzed using techniques such as analytical ultracentrifugation (AUC) or multi-angle-laser-light-scattering (MALLS) for biological interpretation.
Scatter IV is a new, software package, free for research and industrial use with an intuitive user interface that allows even non-experts to analyze their data. Scatter IV has several new features that help to improve the analysis of SEC-SAXS data, such as the heat map linked to the signal plot, enabling greater accuracy with choice of frame selection. In primary data analysis, the Guinier Peak analysis and the cross-validation plot associated with the P(r) analysis offer an integrated troubleshooting ability in the software.
It should be mentioned that many other programs can be used for primary data analysis; these contain the same basic features and are also updated regularly such as BioXTAS RAW17 ATSAS package24 and US-SOMO15 to name a few.
But regardless of which SAXS package is used for analysis, the major limitations are common: the sample preparation, before collection and analysis. In the E9 exominus example shown, it is clear to see the improvement in the signal to noise ratio and with a reduction in the Rg the dmax associated with a monodisperse sample. This will greatly aid further processing of the data such as fitting or modeling with known high-resolution structures.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
We acknowledge the financial support for the project from the French grant REPLIPOX ANR-13-BSV8-0014 and by research grants from the Service de Santé des Armées and the Délégation Générale pour l'Armement. We are thankful to the ESRF for the SAXS beam time. This work used the platforms of the Grenoble Instruct-ERIC center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-05-02) and GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). IBS acknowledges integration into the Interdisciplinary Research Institute of Grenoble (IRIG, CEA). We thank Wim P. Burmeister and Frédéric Iseni for financial and scientific support and we also thank Dr. Jesse Hopkins from BioCAT at the APS for his help and for developing BioXTAS RAW.
Materials
| Beamline control software BsXCuBE | ESRF | Pernot et al. (2013), J. Synchrotron Rad. 20, 660-664 | local development |
| BioXTAS Raw 1.2.3. | MacCHESS | http://bioxtas-raw.readthedocs.io/en/latest/index.html | First developed in 2008 by Soren Skou as part of the biological x-ray total analysis system (BioXTAS) project. Since then it has been extensively developed, with recent work being done by Jesse B. Hopkins |
| HPLC program LabSolutions | Shimadzu | n.a. | |
| ISPyB | ESRF | De Maria Antolinos et al. (2015). Acta Cryst. D71, 76-85. | local development |
| NaCl | VWR Chemicals (BDH Prolabo) | 27808.297 | |
| Scatter | Diamond Light Source Ltd | http://www.bioisis.net/tutorial/9 | Supported by SIBYLS beamline (ALS berkeley, Ca) and Bruker Cororation (Karlsruhe, Germany) |
| Superdex 200 Increase 5/150 GL column | GE Healthcare | 28990945 | SEC-SAXS column used |
| Tris base | Euromedex | 26-128-3094-B |
Referenzen
- Pelikan, M., Hura, G., Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. General Physiology and Biophysics. 28 (2), 174-189 (2009).
- Brosey, C. A., Tainer, J. A. Evolving SAXS versatility: solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Current Opinion in Structural Biology. 58, 197-213 (2019).
- Gräwert, M., Svergun, D. A beginner’s guide to solution small-angle X-ray scattering (SAXS). The Biochemist. 42 (1), 36-42 (2020).
- Putnam, C. D., Hammel, M., Hura, G. L., Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly reviews of biophysics. 40 (03), 191-285 (2007).
- Meisburger, S. P., et al. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. Journal of the American Chemical Society. 138 (20), 6506-6516 (2016).
- Brennich, M. E., Round, A. R., Hutin, S. Online Size-exclusion and Ion-exchange Chromatography on a SAXS Beamline. Journal of Visualized Experiments. (119), e54861 (2017).
- Watanabe, Y., Inoko, Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. Journal of Chromatography A. 1216 (44), 7461-7465 (2009).
- Graewert, M. A., et al. Automated Pipeline for Purification, Biophysical and X-Ray Analysis of Biomacromolecular Solutions. Scientific reports. 5, (2015).
- David, G., Pérez, J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. Journal of applied crystallography. 42 (5), 892-900 (2009).
- Ryan, T. M., et al. An optimized SEC-SAXS system enabling high X-ray dose for rapid SAXS assessment with correlated UV measurements for biomolecular structure analysis. Journal of Applied Crystallography. 51 (1), 97-111 (2018).
- Gampp, H., Maeder, M., Meyer, C. J., Zuberbühler, A. D. Calculation of equilibrium constants from multiwavelength spectroscopic data-III: Model-free analysis of spectrophotometric and ESR titrations. Talanta. 32 (12), 1133-1139 (1985).
- Maeder, M., Neuhold, Y. M. . Practical Data Analysis in Chemistry. , (2007).
- Tarbouriech, N., et al. The vaccinia virus DNA polymerase structure provides insights into the mode of processivity factor binding. Nature Communications. 8 (1), (2017).
- Brennich, M. E., et al. Online data analysis at the ESRF bioSAXS beamline, BM29. Journal of Applied Crystallography. 49 (1), (2016).
- Brookes, E., Rocco, M. Recent advances in the UltraScan SOlution MOdeller (US-SOMO) hydrodynamic and small-angle scattering data analysis and simulation suite. European Biophysics Journal. 47 (7), 855-864 (2018).
- Malaby, A. W., et al. Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering. Journal of Applied Crystallography. 48 (4), 1102-1113 (2015).
- Hopkins, J. B., Gillilan, R. E., Skou, S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. Journal of Applied Crystallography. 50 (5), 1545-1553 (2017).
- Maeder, M. Evolving factor analysis for the resolution of overlapping chromatographic peaks. Analytical Chemistry. 59 (3), 527-530 (1987).
- Durand, D., et al. NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. Journal of Structural Biology. 169 (1), 45-53 (2010).
- De Maria Antolinos, A., et al. ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta Crystallographica Section D. 71 (1), 76-85 (2015).
- Brennich, M. E., et al. Online data analysis at the ESRF bioSAXS beamline, BM29. Journal of Applied Crystallography. 49 (1), 203-212 (2016).
- Kirby, N., et al. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallographica Section D Structural Biology. 72 (12), 1254-1266 (2016).
- Rambo, R. P., Tainer, J. A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 95 (8), 559-571 (2011).
- Franke, D., et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography. 50 (4), 1212-1225 (2017).
