Described herein is a C. elegans workflow that includes culturing, image acquisition, and processing protocols that allow assessment of polyQ aggregation in the presence of various bacteria using a 24-well plate format as the culturing and imaging platform (Figure 1). This protocol can be adjusted to study the effect of bacteria, specific conditions, small molecules, drugs, or genomic manipulations on host proteostasis. The described method has been optimized using worms that constitutively express intestinal polyQ fused to a yellow fluorescent protein (vha6p::polyQ44::YFP); however, other models that report on proteostasis in muscle or neurons can also be used with further optimization. For example, preliminary experiments demonstrate the application of these methods in the quantification of protein aggregates in other tissues such as muscle polyQ (Supplemental Figure 1). However, modification to the pipeline will be required to properly adjust for aggregate size and brightness, as mentioned in section 8 NOTE.
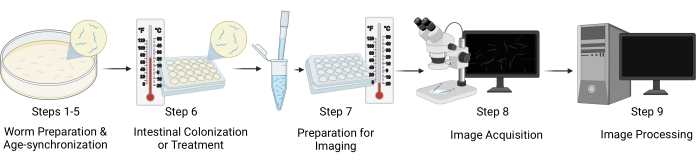
Figure 1: Workflow visual representation. The major steps of the protocol include five distinct stages: worm preparation and age-synchronization (steps 1-5), intestinal colonization/worm treatment (step 6), sample preparation for imaging (step 7), image acquisition (step 8), and image processing (step 9). The "Sections" of the protocol are referenced as "Steps" in the figure. Please click here to view a larger version of this figure.
The initial optimization experiments revealed various difficulties associated with overcrowding due to a large number of progenies, resulting in faster food depletion. The supplementation of FUDR in NGM plates described in section 2 solved this problem (Figure 2). Additionally, in the presence of FUDR, worms that were fed various bacteria had a more consistent body size, which allowed more uniform and accurate worm detection.
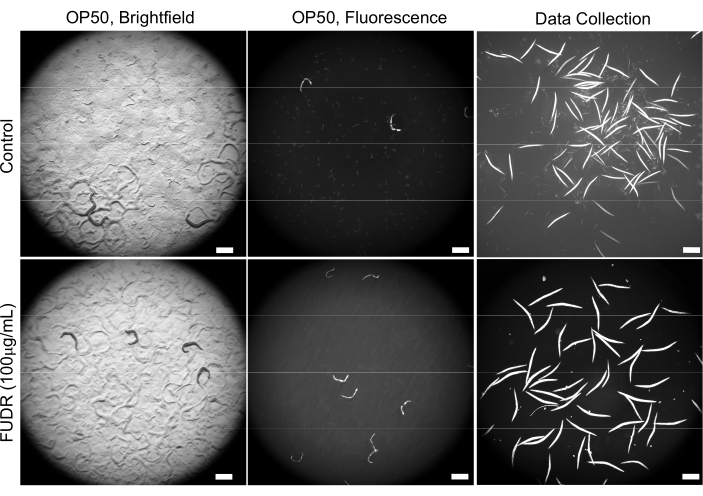
Figure 2: The use of FUDR improves image quality by reducing progeny. FUDR-supplemented plates eliminate C. elegans progeny compared to worms grown on non-FUDR control NGM plates seeded with E. coli OP50. Images were acquired at 25.2x magnification (40x magnification with a 0.63x camera adapter). Scale bars = 500 µm. Please click here to view a larger version of this figure.
Background fluorescence contributed to false positive detection of polyQ aggregates. To reduce such fluorescence signal in the intestinal tract and improve automated detection of aggregates, it was necessary to freeze worms prior to imaging. Freezing worms at -20 °C for 18-48 h significantly improved polyQ aggregate detection by eliminating background fluorescence (Figure 3A). The human eye is capable of differentiating between aggregates and background fluorescence; hence the manual counting before and after the freeze is the same (Figure 3B). However, automated counting is not as accurate, but freezing significantly improved automated counts with accuracy comparable to manual counting (Figure 3B).
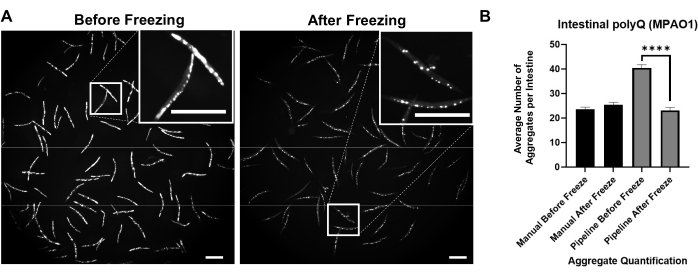
Figure 3: Freezing improves aggregate detection. (A) Fluorescent images of C. elegans expressing intestinal polyQ44::YFP before and after freezing. Inserts represent close-up images of the selected area. Scale bars = 500 µm. (B) Average number of aggregates per intestine in worms colonized with P. aeruginosa MPAO1 before and after freezing using manual or automated (pipeline) aggregate quantification. Data represent two biological replicates (n = 60-109). Statistical significance was calculated using Student's t-test (**** p < 0.0001). Error bars represent the standard error of the mean (SEM). Please click here to view a larger version of this figure.
Inverted brightfield illumination was used to detect the whole C. elegans (Figure 4A) and GFP channel to image polyQ44::YFP aggregates (Figure 4B). Worm detection, detangling, and aggregate quantification for each worm were done by applying an optimized CellProfiler image processing pipeline (Supplemental File 1), which allows obtaining the number of aggregates per individual worm (Figure 4C-D). To test the feasibility of this approach and the accuracy of the automated aggregate detection and quantification, worms expressing intestine-specific polyQ44::YFP were cultured and prepared for imaging according to the established protocols (sections 1-7). The number of aggregates per worm was assessed using either the automated pipeline (sections 8-9) or manual counting. Each experiment was performed in three independent trials using 90-571 worms per condition. While the average number of aggregates per intestine obtained with two trials had no significant difference, worms in the third trial had significantly fewer aggregates when quantified using the automated approach (Figure 5A). The average number of aggregates from the three trials resulted in slightly, but significantly fewer aggregates when the quantification was done using the CellProfiler pipeline (sections 8-9) (Figure 5B). Nonetheless, the difference between the two approaches was minimal, indicating that the automated method can be applied to large-scale screens.
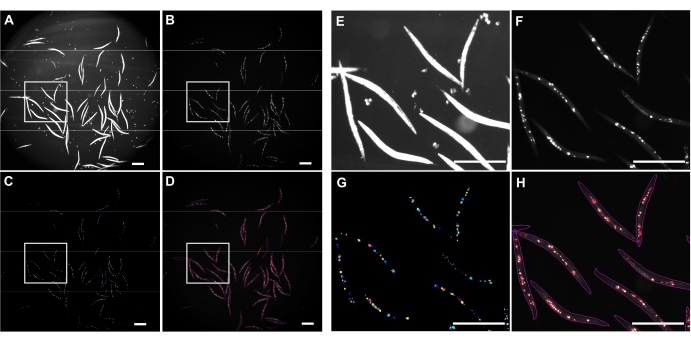
Figure 4: Aggregate detection using CellProfiler. (A) Brightfield image used to identify worm bodies. (B) Original fluorescent image acquired using GFP channel and used to identify and quantify a total number of intestinal polyQ44::YFP aggregates. (C) Aggregates identified using CellProfiler. (D) A total number of identified aggregates superimposed over the original fluorescent image with worm and aggregate outlines. Image capture and processing were performed using the settings described in sections 8-9. Panels E–H represent close-up images of the corresponding outlined regions in images A–D. Images were acquired at 25.2x magnification (40x magnification with a 0.63x camera adapter). Scale bars = 500 µm. Please click here to view a larger version of this figure.
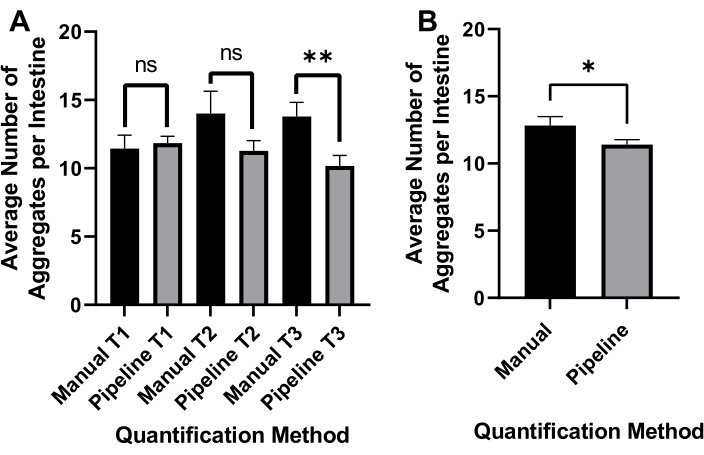
Figure 5: Efficacy of automated aggregate quantification. (A) Average aggregate number per intestine in worms colonized with control E. coli OP50 using manual counting (Manual) and automated CellProfiler-based quantification (Pipeline). Results represent data analyzed in three separate trials (T1-T3) (n = 90-571). (B) The average number of aggregates per intestine was obtained using manual or automated (Pipeline) aggregate quantification. Statistical significance was calculated using Student's t-test (* p < 0.05, ** p < 0.01). Error bars represent SEM. Please click here to view a larger version of this figure.
To evaluate the reproducibility of results among different experimenters, images from a plate containing six wells of worms that were fed either Pseudomonas aeruginosa MPAO1 or Escherichia coli OP50, were acquired by three individuals, two of whom had no prior experience imaging worms using these protocols. Images collected from each well contained anywhere between 30-115 detected worms. A non-significant difference in aggregation was detected in worms from the same wells that were imaged by the three experimenters. While the average number of aggregates per intestine remained very consistent between the three experimenters for worms fed MPAO1 and OP50, there were some statistically significant differences in the average number of aggregates but only in worms colonized by MPAO1 (Supplemental Figure 2). These results highlight the reproducibility of the results even between inexperienced experimenters.
To ensure that the reproducibility of aggregate quantification is not significantly influenced by worm position, a set of 15 worms was selected and imaged 15 separate times following agitation in between each image capture using a pipette tip. Images of aggregates in worms fed with E. coli OP50 and P. aeruginosa MPAO1 were collected and analyzed using CellProfiler. The average number of aggregates from each of these different sets of images was slightly but non-significantly different, further supporting the reproducibility of this approach (Supplemental Figure 3).
Colonization of the C. elegans intestine with gram-negative enteric pathogens has been shown to disrupt proteostasis across tissues, with P. aeruginosa being among the most potent inducers of polyQ aggregation9. To determine whether these optimized protocols will successfully detect and quantify P. aeruginosa-mediated enhancement of aggregation, worms expressing intestinal polyQ were colonized with E. coli OP50 (control bacteria), and P. aeruginosa MPAO1, sections 1-8 were conducted. The acquired images were analyzed using CellProfiler (section 9, Supplemental File 1). The results of automated quantification show a significant increase in the number of aggregates induced by P. aeruginosa MPAO1, consistently resulting in a two-fold enhancement compared to worms fed with control E. coli OP50 (Figure 6).
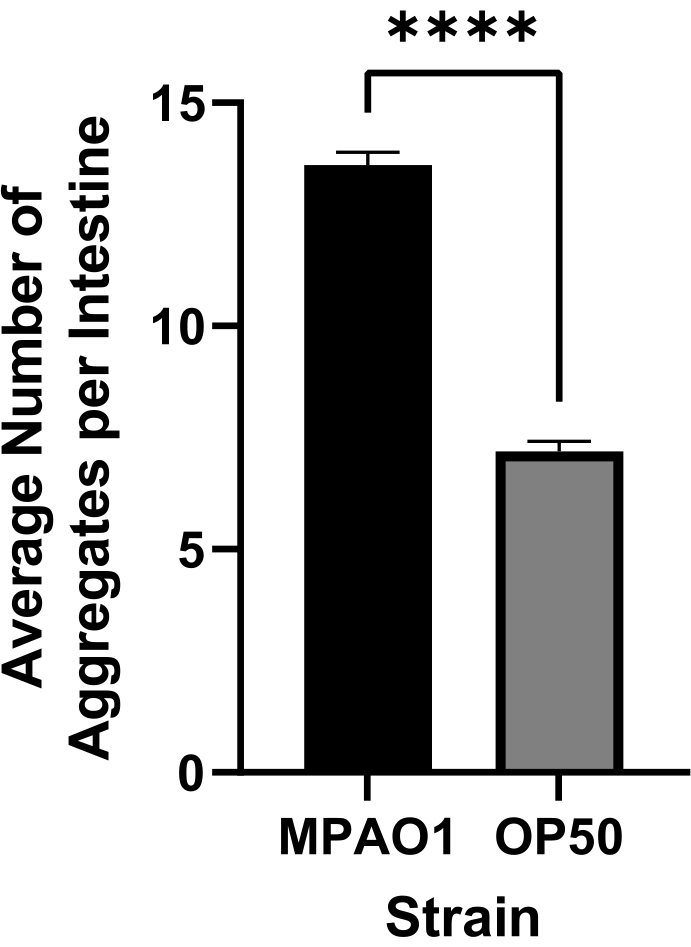
Figure 6: The average number of aggregates per intestine in worms colonized with control E. coli OP50 and P. aeruginosa MPAO1. The number of aggregates per intestine was assessed using CellProfiler (sections 8-9). Data are represented as the average number of aggregates per intestine in worms colonized with OP50 (n = 1068) and MPAO1 (n = 1557). Statistical significance was calculated using Student's t-test (**** p < 0.0001). Error bars represent SEM. Please click here to view a larger version of this figure.
The optimized pipeline was designed to support large-scale screens for conditions that affect proteostasis. To test the feasibility of this approach in screening large libraries of bacteria for their effect on host proteostasis, the pipeline described herein was employed (sections 1-9) to test the effect of 90 P. aeruginosa non-essential gene knock-out mutant strains on polyQ aggregation18. This pilot screen is part of a larger project designed to screen all P. aeruginosa non-essential mutant strains for their ability to influence host proteostasis. Out of 90 bacterial strains tested, colonization of C. elegans intestine with one candidate showed a significant decrease in the number of aggregates (Figure 7). Follow-up experiments to evaluate the sensitivity of this assay were performed via manual aggregate counts from a random selection of six P. aeruginosa mutants that differed non-significantly from the MPAO1 control. These experiments were performed using the more traditional 6 cm NGM plates, transferring worms onto test strains as L1's to recapitulate previously established methods9. The confirmation experiments by manual counting revealed that none of the mutants, including the one that significantly decreased the number of aggregates (Figure 7), affected polyQ aggregation (Supplemental Figure 4). In addition, the subtle changes in aggregation observed in the screen of the 90 mutant strains were not detected among manual counts of selected candidates, indicating that such changes could arise because of biological and experimental variability, such as low n value. Collectively, the results indicate that while our method can reliably pick up significant changes, subtle ones will likely be missed, and all potential candidates will have to be individually confirmed.
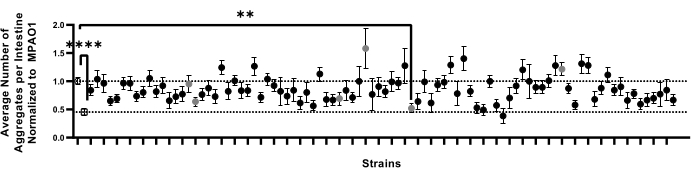
Figure 7: The number of aggregates per intestine in a representative sample set of worms colonized by 92 bacterial strains. Data are represented as the average number of aggregates per intestine normalized to that of worms colonized with MPAO1. Dotted lines represent the average number of aggregates in worms colonized with MPAO1 (top, open circle) and OP50 control (bottom, open square). Solid symbols represent 90 distinct knock-out mutant strains of P. aeruginosa MPAO1. The average number of aggregates per worm between worms colonized with MPAO1 and a single mutant was statistically significant. Gray circles represent samples that were confirmed manually (Supplemental Figure 4). Statistical significance was calculated using one-way analysis of variance (ANOVA) followed by multiple comparison Dunnett's post-hoc test (** p < 0.01, **** p < 0.0001). Error bars represent SEM. Please click here to view a larger version of this figure.
To manage the large amount of data generated by CellProfiler, a Graphical User Interface (GUI) was developed to automate data processing and organization (Figure 8). The GUI was developed using Tkinter, an open-source Python cross-platform widget toolkit. From the given metadata, the application extracts the number of aggregates (column K) from each well (Column J) present in a plate. A Python data handling library called "Pandas" was used to carry out the aforementioned process. The GUI application provides drag-and-drop support for users to upload data files. The data in each file are stored in the form of a two-dimensional tabular structure called a data frame. An empty dictionary pair is initialized for every unique well found within the data frame. Next, the distinct aggregates found in each well are counted and appended to their respective dictionary pairs. The column with lesser data is padded with empty valued strings to ensure that each column is even in size. Finally, the structure is converted to a data frame which is exported in the form of a spreadsheet into the directory specified by the user.
Figure 8: Graphical User Interface. Please click here to view a larger version of this figure.
Supplemental Figure 1: Detection of muscle-specific polyQ aggregates. Worms expressing muscle-specific polyQ35::YFP were plated as L1s and cultured on OP50 for 48 h. Once worms developed into young adults, they were transferred to 24-well NGM plates, supplemented with 100 µg/mL FUDR and seeded with MPAO1 for an additional 72 h before imaging. (A) Brightfield image used to identify worm bodies. (B) Original fluorescent image acquired using GFP channel. (C) Aggregates identified using CellProfiler. (D) A total number of identified aggregates superimposed over the original fluorescent image with worm and aggregate outlines. Image capture and processing were performed using the settings described in sections 8-9. Panels E–H represent close-up images of the corresponding outlined regions in images A–D. Scale bars = 500 µm. Please click here to download this File.
Supplemental Figure 2: Reproducibility of aggregate quantification between different experimenters. Average number of aggregates quantified using CellProfiler for six wells of worms colonized with P. aeruginosa MPAO1 (black bars) and six wells of worms colonized with control E. coli OP50 (gray bars). Each well was imaged by three experimenters (AVS, DMC, RDH). Data are represented as the average number of aggregates per intestine (n = 30-115). Statistical significance was calculated using one-way ANOVA followed by Tukey's multiple comparisons test (* p < 0.05, ** p < 0.01). Error bars represent SEM. Please click here to download this File.
Supplemental Figure 3: The effect of worm position on the reproducibility of aggregate quantification. Average aggregate number per intestine in worms colonized with control E. coli OP50 (gray bars) and P. aeruginosa MPAO1 (black bars). Results represent the average number of aggregates per intestine (15≥n≥12) quantified using CellProfiler. The position of worms within the wells was changed by agitation in between each acquisition. No statistically significant differences were found in either group. Statistical significance was calculated using one-way ANOVA followed by Tukey's multiple comparisons test. Error bars represent SEM. Please click here to download this File.
Supplemental Figure 4: Confirmation of the pilot screen with manual counting. The average number of aggregates per intestine was quantified manually. Data represent aggregation profiles of worms colonized with six MPAO1 knock-out mutants (gray circles Figure 7) compared against wild-type MPAO1 and OP50 controls (n = 30). Statistical significance was calculated using one-way ANOVA followed by multiple comparison Dunnett's post-hoc test (**** p < 0.0001). Error bars represent SEM. Please click here to download this File.
Supplemental Figure 5: The effect of FUDR on intestinal polyQ aggregation. Data are represented as the average number of polyQ44::YFP aggregates per intestine (n = 20). Worms were transferred onto control (no FUDR) or FUDR-containing plates (100 µg/mL) after 48 h of growth at 25 °C on E. coli OP50. Manual counts were collected after an additional 48 h. Statistical significance was calculated using Student's t-test (ns = not significant). Error bars represent SEM. Please click here to download this File.
Supplemental File 1: Proteostasis pipeline. Downloadable image analysis pipeline for use in CellProfiler. Instructions for application can be found in section 9. Please click here to download this File.
Supplemental File 2: Training set untangling worm. File to be uploaded into the "UntangleWorms" module. This particular training set is specific to the worms used in the initial approach. Alterations in worm size and shape will change the accuracy and quality of identification. It may be necessary to create a more personalized training file. Instructions for creating a new training set can be found at the official CellProfiler website17. Please click here to download this File.
Supplemental File 3: Graphical User Interface for Windows Operating System. gui_windowsOS_64x.zip. Please click here to download this File.
Supplemental File 4: Graphical User Interface for Mac Operating System. gui_MacOS_64x.zip. Please click here to download this File.
