Chromosoomconformatie vastleggen over lengteschalen
Summary
Hi-C 3.0 is een verbeterd Hi-C protocol dat formaldehyde en disuccinimidylglutaraat crosslinkers combineert met een cocktail van DpnII en DdeI restrictie enzymen om de signaal-ruisverhouding en de resolutie van chromatine interactie detectie te verhogen.
Abstract
Chromosoom conformatie capture (3C) wordt gebruikt om driedimensionale chromatine interacties te detecteren. Meestal wordt chemische crosslinking met formaldehyde (FA) gebruikt om chromatine-interacties te fixeren. Vervolgens zet chromatinevertering met een restrictie-enzym en daaropvolgende religatie van fragmenteinden driedimensionale (3D) nabijheid om in unieke ligatieproducten. Ten slotte, na omkering van crosslinks, eiwitverwijdering en DNA-isolatie, wordt DNA geschoren en voorbereid voor high-throughput sequencing. De frequentie van nabijheidsligatie van paren loci is een maat voor de frequentie van hun colocalisatie in de driedimensionale ruimte in een celpopulatie.
Een gesequencede Hi-C-bibliotheek biedt genoombrede informatie over interactiefrequenties tussen alle paren loci. De resolutie en precisie van Hi-C is afhankelijk van efficiënte crosslinking die chromatinecontacten en frequente en uniforme fragmentatie van het chromatine handhaaft. Dit artikel beschrijft een verbeterd in situ Hi-C-protocol, Hi-C 3.0, dat de efficiëntie van crosslinking verhoogt door twee crosslinkers (formaldehyde [FA] en disuccinimidylgluutaraat [DSG]) te combineren, gevolgd door een fijnere spijsvertering met behulp van twee restrictie-enzymen (DpnII en DdeI). Hi-C 3.0 is een enkel protocol voor de nauwkeurige kwantificering van genoomvouwingskenmerken op kleinere schalen zoals lussen en topologisch associërende domeinen (TAD’s), evenals functies op grotere kernbrede schalen zoals compartimenten.
Introduction
Sinds 2002 wordt chromosoomconformatie-opname gebruikt1. Fundamenteel is elke conformatievangstvariant afhankelijk van de fixatie van DNA-eiwit- en eiwit-eiwitinteracties om de 3D-chromatine-organisatie te behouden. Dit wordt gevolgd door DNA-fragmentatie, meestal door restrictievertering, en ten slotte religatie van nabijgelegen DNA-uiteinden om ruimtelijk proximale loci om te zetten in unieke covalente DNA-sequenties. Initiële 3C-protocollen gebruikten PCR om specifieke, “één-op-één” interacties te bemonsteren. Latere 4C-assays maakten de detectie van “one-to-all” interacties2 mogelijk, terwijl 5C “many-to-many” interacties detecteerde3. Chromosoomconformatie-opname kwam tot volle wasdom na de implementatie van next-generation, high-throughput sequencing (NGS), die detectie van “all-to-all” genomische interacties mogelijk maakte met behulp van genoombrede Hi-C4 en vergelijkbare technieken zoals 3C-seq5, TCC6 en Micro-C 7,8 (zie ook review door Denker en De Laat9).
In Hi-C worden gebiotinyleerde nucleotiden gebruikt om 5′-overhangen na de spijsvertering en vóór ligatie te markeren (figuur 1). Dit maakt de selectie van goed verteerde en religated fragmenten mogelijk met behulp van streptavidin-gecoate kralen, waardoor het zich onderscheidt van GCC10. Een belangrijke update van het Hi-C-protocol werd geïmplementeerd door Rao et al.11, die de spijsvertering en religatie in intacte kernen (d.w.z. in situ) uitvoerden om valse ligatieproducten te verminderen. Bovendien verminderde het vervangen van HindIII-spijsvertering door MboI (of DpnII) -spijsvertering de fragmentgrootte en verhoogde het het resolutiepotentieel van Hi-C. Deze toename maakte de detectie van relatief kleinschalige structuren en een nauwkeurigere genomische lokalisatie van contactpunten mogelijk, zoals DNA-lussen tussen kleine cis-elementen, bijvoorbeeld lussen tussen CTCF-gebonden locaties gegenereerd door lusextrusie11,12. Dit potentieel brengt echter kosten met zich mee. Ten eerste vereist een tweevoudige toename van de resolutie een viervoudige (22) toename van de sequencingreads 13. Ten tweede vergroten de kleine fragmentgroottes de kans om onverteerde naburige fragmenten te verwarren met verteerde en religated fragmenten14. Zoals vermeld, verschillen in Hi-C verteerde en religated fragmenten van onverteerde fragmenten door de aanwezigheid van biotine op de ligatieverbinding. Een goede verwijdering van biotine uit niet-geliformeerde uiteinden is echter vereist om ervoor te zorgen dat alleen ligatieverbindingen14,15 naar beneden worden getrokken.
Met de dalende kosten van NGS wordt het haalbaar om chromosoomvouwing in meer detail te bestuderen. Om de grootte van DNA-fragmenten te verkleinen en daardoor de resolutie te verhogen, kan het Hi-C-protocol worden aangepast om vaker snijdende restrictie-enzymen16 te gebruiken of om combinaties van restrictie-enzymen te gebruiken 17,18,19. Als alternatief kunnen MNase 7,8 in Micro-C en DNase in DNase Hi-C20 worden getitreerd om een optimale spijsvertering te bereiken.
Een recente systematische evaluatie van de fundamenten van 3C-methoden toonde aan dat de detectie van chromosoomvouwingskenmerken op elke lengteschaal sterk verbeterde met sequentiële crosslinking met 1% FA gevolgd door 3 mM DSG17. Bovendien was Hi-C met HindIII-spijsvertering de beste optie voor het detecteren van grootschalige vouwfuncties, zoals compartimenten, en dat Micro-C superieur was in het detecteren van kleinschalige vouwfuncties zoals DNA-lussen. Deze resultaten leidden tot de ontwikkeling van een enkele, hoge resolutie “Hi-C 3.0” strategie, die de combinatie van FA en DSG crosslinkers gebruikt, gevolgd door dubbele vertering met DpnII en DdeI endonucleases21. Hi-C 3.0 biedt een effectieve strategie voor algemeen gebruik omdat het nauwkeurig vouwfuncties detecteert op alle lengteschalen17. Het experimentele gedeelte van het Hi-C 3.0-protocol wordt hier gedetailleerd en typische resultaten die na sequencing kunnen worden verwacht, worden weergegeven.
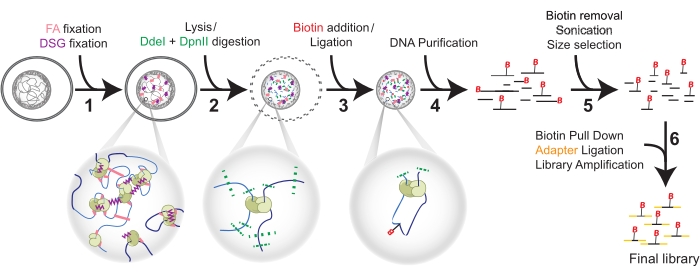
Figuur 1: Hi-C procedure in zes stappen. Cellen worden eerst gefixeerd met FA en vervolgens met DSG (1). Vervolgens gaat lysis vooraf aan een dubbele spijsvertering met DdeI en DpnII (2). Biotine wordt toegevoegd door overhang fill-in en proximale stompe uiteinden worden geligeerd (3) vóór DNA-zuivering (4). Biotine wordt verwijderd uit niet-geliformeerde uiteinden vóór ultrasoonapparaat en grootteselectie (5). Ten slotte maakt het naar beneden trekken van biotine adapterligatie en bibliotheekversterking door PCR mogelijk (6). Afkortingen: FA = formaldehyde; DSG = disuccinimidylglutaraat; B = Biotine. Klik hier om een grotere versie van deze figuur te bekijken.
Protocol
Representative Results
Discussion
Kritieke stappen voor celverwerking
Hoewel het mogelijk is om een lager aantal invoercellen te gebruiken, is dit protocol geoptimaliseerd voor ~ 5 × 106 cellen per sequencing lane (~ 400 M reads) om de juiste complexiteit na diepe sequencing te garanderen. Cellen kunnen het beste worden geteld voorafgaand aan fixatie. Voor het genereren van ultradeep bibliotheken vermenigvuldigen we over het algemeen het aantal lanes (en cellen) totdat de gewenste leesdiepte is bereikt. Voor een optimale fixatie moet serumhoudend medium vóór FA-fixatie worden vervangen door PBS en moeten fixatieve oplossingen onmiddellijk en zonder concentratiegradiënten15,22 worden toegevoegd. Voor het oogsten van cellen heeft schrapen de voorkeur boven trypsinisatie, omdat de overgang van een plattere naar een bolvorm na trypsinisatie de nucleaire conformatie kan beïnvloeden. Na de toevoeging van DSG gaan losse en klonterige celpellets gemakkelijk verloren. Wees voorzichtig bij het hanteren van cellen in dit stadium en voeg tot 0,05% BSA toe om klonteren te verminderen.
Wijzigingen in de methode
Dit protocol is ontwikkeld met behulp van menselijke cellen17. Maar op basis van ervaring met het vastleggen van chromosoomconformaties zou dit protocol voor de meeste eukaryote cellen moeten werken. Voor een significant lagere input (~1 × 106 cellen) adviseren wij de helft van de volumes te gebruiken voor de lysis- en conformatievangstprocedures [stappen 2.1-2.4]. Hierdoor zou DNA-isolatie [stap 2.5] ook kunnen worden uitgevoerd in een tafelcentrifuge met buizen van 1,7 ml, wat de pelletering voor lage DNA-concentraties zou kunnen verbeteren. De kwantificering van DNA (stap 2.6.6) geeft aan hoe verder te gaan. Voor lage hoeveelheden geïsoleerd DNA (1-5 μg) raden we aan de grootteselectie over te slaan (stap 3.3) en door te gaan met het verwijderen van biotine na het verminderen van het volume van 130 μL tot ~ 45 μL met een CFU.
Dit protocol is speciaal ontwikkeld om gegevens van hoge kwaliteit te garanderen na daaropvolgende crosslinking met FA en DSG en vertering met DpnII en DdeI. Alternatieve crosslinkingstrategieën zoals FA gevolgd door EGS (ethyleenglycol bis (succinimidylsuccinaat)), die ook wordt gebruikt in ChIP-seq23 en ChIA-PET24, kunnen echter even goed werken17. Evenzo kunnen verschillende enzymcombinaties, zoals DpnII en HinfI18 of MboI, MseI en NlaIII19 , worden gebruikt voor de spijsvertering. Wanneer u enzymcombinaties aanpast, moet u gebiotinyleerde nucleotiden gebruiken die de specifieke 5 ‘overhangen kunnen invullen en de meest optimale buffers voor elke cocktail kunnen gebruiken. DpnII wordt geleverd met een eigen buffer en de enzymfabrikant beveelt een specifieke buffer aan voor DdeI-vertering. Toch wordt voor de dubbele vertering met DpnII en DdeI in dit protocol restrictiebuffer aanbevolen omdat deze voor beide enzymen op 100% activiteit wordt beoordeeld.
Problemen met het vastleggen van conformaties oplossen
De drie belangrijkste stappen in het vastleggen van chromosoomconformatie: crosslinking, spijsvertering en religatie zijn allemaal uitgevoerd voordat de resultaten op gel kunnen worden gevisualiseerd. Om de kwaliteit van elk van deze drie stappen te bepalen en te onderscheiden waar problemen kunnen zijn ontstaan, worden aliquots voor (CI) en na de spijsvertering (DC) genomen en samen met het geligeerde Hi-C-monster op de gel geladen (figuur 2). Deze gel wordt gebruikt om de kwaliteit van het Hi-C-monster te bepalen en of het de moeite waard is om het protocol voort te zetten. Zonder het CI en DC is het moeilijk om potentiële suboptimale stap(en) aan te wijzen. Het is vermeldenswaard dat suboptimale ligatie te wijten kan zijn aan een probleem in de ligatie zelf, de invulling of een probleem met crosslinking. Om problemen met crosslinking op te lossen, moet u niet meer dan 1 × 107 cellen per bibliotheek gebruiken en beginnen met nieuwe crosslinking-reagentia en schone cellen (d.w.z. gespoeld met PBS). Zorg er voor ligatie voor dat cellen en ligatiemengsels op ijs worden gehouden. Voeg T4 DNA ligase toe vlak voor de 4 uur incubatie bij 16 °C en meng goed.
Problemen met bibliotheekvoorbereiding oplossen
Als er meer dan 10 PCR-cycli nodig zijn of als er geen PCR-product op gel te zien is na PCR-titratie (figuur 4), zijn er een paar opties om het Hi-C-monster op te slaan. Terugwerkend van de PCR-titratie, is de eerste optie om de PCR opnieuw te proberen. Als er nog steeds niet genoeg product is, is het mogelijk om nog een ronde A-tailing en adapterligatie (stap 3.6) te proberen na het tweemaal wassen van de kralen met 1x TLE-buffer. Na deze extra A-tailing en adapterligatie kan men overgaan tot de PCR-titratie zoals voorheen. Als er nog steeds geen product is, is de laatste optie om de 0,8x-fractie uit stap 3.3 te resoneren en van daaruit verder te gaan.
Beperkingen en voordelen van Hi-C3.0
Het is belangrijk om te beseffen dat Hi-C een populatie-gebaseerde methode is die de gemiddelde frequentie van interacties tussen paren loci in de celpopulatie vastlegt. Sommige computationele analyses zijn ontworpen om combinaties van conformaties van een populatie van25 te ontwarren, maar in principe is Hi-C blind voor verschillen tussen cellen. Hoewel het mogelijk is om eencellige Hi-C26,27 uit te voeren en computationele gevolgtrekkingen kunnen worden gemaakt28, is hi-c met één cel niet geschikt voor het verkrijgen van 3C-informatie met ultrahoge resolutie. Een extra beperking van Hi-C is dat het alleen paarsgewijze interacties detecteert. Om multicontactinteracties te detecteren, kan men frequente cutters gebruiken in combinatie met short-read sequencing (Illumina)16 of multicontact 3C29 of 4C30 uitvoeren, met behulp van long-read sequencing van PacBio- of Oxford Nanopore-platforms. Hi-C-derivaten om specifiek contacten tussen en langs zusterchromatiden te detecteren zijn ook ontwikkeld 31,32.
Hoewel Hi-C19 en Micro-C33 kunnen worden gebruikt om contactkaarten te genereren met subkilobase-resoluties, vereisen beide een grote hoeveelheid sequencing-reads en dit kan een kostbare onderneming worden. Om zonder de kosten tot een vergelijkbare of zelfs hogere resolutie te komen, kan verrijking voor specifieke genomische regio’s (capture-C34) of specifieke eiwitinteracties (ChiA-PET35, PLAC-seq36, Hi-ChIP37) worden toegepast. De kracht en het nadeel van deze verrijkingstoepassingen is dat slechts een beperkt aantal interacties wordt bemonsterd. Met dergelijke verrijkingen gaat het mondiale aspect van Hi-C (en de optie van wereldwijde normalisatie) verloren.
Belang en potentiële toepassingen van Hi-C3.0
Dit protocol is ontworpen om ultradeeps 3C met hoge resolutie mogelijk te maken en tegelijkertijd grootschalige vouwfuncties zoals TAD’s en compartimenten17 te detecteren (figuur 6). Dit protocol begint met 5 × 106 cellen per buis voor elke Hi-C-bibliotheek, wat meer dan genoeg materiaal zou moeten zijn om een of twee banen op een stroomcel te sequencen om tot 1 miljard gepaarde eindwaarden te verkrijgen. Voor ultradeep sequencing moeten meerdere buizen van 5 × 106 cellen worden voorbereid, afhankelijk van het aantal toegewezen reads en PCR-duplicaten. Bij de hoogste resolutie (<1 kb) worden looping-interacties meestal gevonden tussen CTCF-sites, maar promotor-enhancer-interacties kunnen ook worden gedetecteerd. Lezers kunnen akgol Oksuz et al.17 raadplegen voor een gedetailleerde beschrijving van de data-analyse.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
We willen Denis Lafontaine bedanken voor de ontwikkeling van protocollen en Sergey Venev voor bio-informaticahulp. Dit werk werd ondersteund door een subsidie van het National Institutes of Health Common Fund 4D Nucleome Program aan J.D. (U54-DK107980, UM1-HG011536). J.D. is een onderzoeker van het Howard Hughes Medical Institute.
Dit artikel is onderworpen aan HHMI’s Open Access to Publications beleid. HHMI-labhoofden hebben eerder een niet-exclusieve CC BY 4.0-licentie aan het publiek en een sublicentieerbare licentie aan HHMI verleend in hun onderzoeksartikelen. Op grond van deze licenties kan het door de auteur geaccepteerde manuscript van dit artikel onmiddellijk na publicatie vrij beschikbaar worden gesteld onder een CC BY 4.0-licentie.
Materials
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3–>5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Referenzen
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -. H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273 (2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198 (2021).
- Belton, J. -. M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174 (2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369 (2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
