A Protein Suspension-Trapping Sample Preparation for Tear Proteomics by Liquid Chromatography-Tandem Mass Spectrometry
Summary
This protocol describes a method for collecting tear samples using Schirmer strips and an integrated quantitative workflow for discovering non-invasive tear protein biomarkers. The suspension trapping sample preparation workflow enables fast and robust tear sample preparation and mass spectrometric analysis, resulting in higher peptide recovery yields and protein identification than standard in-solution procedures.
Abstract
Tear fluid is one of the easily accessible biofluids that can be collected non-invasively. Tear proteomics has the potential to discover biomarkers for several ocular diseases and conditions. The suspension trapping column has been reported to be an efficient and user-friendly sample preparation workflow for the broad application of downstream proteomic analysis. Yet, this strategy has not been well-studied in the analysis of human tear proteome. The present protocol describes an integrated workflow from clinical human tear samples to purified peptides for non-invasive tear protein biomarker research using mass spectrometry, which provides insights into disease biomarkers and monitoring when combined with bioinformatics analysis. A protein suspension trapping sample preparation was applied and demonstrated the discovery of tear proteome with fast, reproducible, and user-friendly procedures, as a universal, optimized sample preparation for human tear fluid analysis. In particular, the suspension trapping procedure outperformed in-solution sample preparation in terms of peptide recovery, protein identification, and shorter sample preparation time.
Introduction
Tear proteomics has received attention to explore potential biomarkers for ocular diseases and conditions1,2,3,4,5,6, to access the pathogenesis of the ocular and systemic condition, as well as to exploit the advantage of non-invasive tear sample collection using Schirmer strips. The technological advancement of next-generation mass spectrometry has enabled protein quantification in microliter-scale tears with accuracy and precision not possible in the past. Sample preparation methods are not yet standardized. A robust and standardized sample preparation workflow is essential for the success of clinical application in tear protein biomarker research. The suspension trapping (S-Trap) sample preparation workflow was recently reported as an effective and sensitive sample preparation method for broad downstream proteomic analysis7,8. Yet, this strategy has not been well-reported in the analysis of human tear proteome, outperformed filter-aided sample preparation (FASP) and in-solution digestion in terms of enzymatic digestion efficiency and the greater number of protein identification by mass spectrometric analysis9. The S-Trap-based approach was demonstrated in the preparation of retinal tissue 10, formalin-fixed, paraffin-embedded (FFPE) tissue11, cells12, microorganism13, and liquid biopsies14,15.
This protocol describes an integrated quantitative workflow from clinical samples to enzymatically digested protein for the discovery of a non-invasive tear protein biomarker panel with a rapid, reproducible, and robust technical strategy. Briefly, tear fluid was collected using a standard Schirmer strip and immediately dried with an ophthalmic frame heater to prevent protein autolysis at room temperature. Embedded total proteins were extracted using 5% sodium dodecyl sulfate (SDS) lysis buffer according to the manufacturer's suggestion, followed by protein assay measurement. The extracted lysate then underwent standard reduction with dithiothreitol (DTT) and alkylation with iodoacetamide (IAA).
After acidification with phosphoric acid, suspension trapping column protein precipitation buffer containing 90% methanol and 100 mM triethylammonium bicarbonate (TEAB) was added to aggregate proteins. The sample was then transferred into a new suspension trapping micro column. Enzymatic digestion with done with sequencing grade trypsin at a 1:25 ratio (w/w, trypsin: protein) at 47 °C for 1 h. The resulting peptides were then eluted via centrifugation, sequentially with 50 mM TEAB, 0.2% aqueous formic acid (FA), and 50% acetonitrile (ACN) containing 0.2% FA. The eluted peptides were vacuum-dried and reconstituted in 0.1 % FA. The peptide concentration was measured and adjusted to 0.5 µg/µL for mass spectrometric analysis.
Protocol
Subjects provided written informed consent before participation in the study. The study was approved by the Human Ethics Committee of The Hong Kong Polytechnic University and adhered to the tenets of the Declaration of Helsinki.
1. Collection of human tear fluid with Schirmer strip
- Wear gloves and sanitize to avoid sample contamination.
- Check that the inner packaging is intact, sterile, and not expired.
- Bend the top of the Schirmer strip inward slightly at the 0 mm mark, as indicated near the semicircle tip of the strip (Figure 1).
- Remove the strip from the inner package by holding the end of the strip, and avoid direct contact with the sample collection area from 0 to 25 mm.
- Ask the subject to look away from the position of the strip to be placed.
- Pull down the lower eyelid gently and hook the end of the strip in the lower fornix near the lateral canthus region (Figure 2). Repeat the same procedure on the other eye.
- Ask the subject to gently close the eyelids to minimize irritation.
- Collect tear fluid for about 5 min or preferably 15-20 mm tear sample was collected.
- Ask the subject to open both eyes, pull down the lower eyelid gently and remove the strips from the subject.
- Inspect the hydrated length on the Schirmer strip and record the collected amount in mm.
- Dry the strip with a standard, clean frame heater until the fluid is completely dry.
NOTE: Excess heating that could potentially char the filter paper should be avoided. - Insert each dried Schirmer strip in a labeled cryotube and store it preferably in a cool, dry place in the dark until further processing.
2. Preparation of chemicals and reagents
- Lysis Buffer (5% SDS, 50 mM TEAB, pH 7.5), 20 mL
- Pipette 1 mL of 1 M TEAB and 80 µL of 12% phosphoric acid. Vortex and sonicate briefly to remove bubbles. Top up to 20 mL with deionized water.
- Weigh 1 g of SDS and add to the solution with oscillation until dissolved.
- Protein Precipitation Buffer (90% methanol, 100 mM TEAB, pH 7.1), 10 mL
- Add 893 µL of 1 M TEAB and 92 µL of deionized water.
- Add 15 µL of 85 wt. % phosphoric acid and 9 mL of methanol to the solution and vortex briefly.
- Digestion Buffer (50 mM TEAB), 1 mL
- Add 50 µL of 1 M TEAB and top up to 1 mL with deionized water.
- 200 mM dithiothreitol (DTT) solution, 500 µL
- Weigh 15 mg of DTT and dissolve in 500 µL of deionized water; vortex briefly.
NOTE: Prepare before use.
- Weigh 15 mg of DTT and dissolve in 500 µL of deionized water; vortex briefly.
- 400 mM iodoacetamide (IAA) solution, 200 µL
- Weigh 15 mg of IAA and dissolve in 200 µL of deionized water; vortex briefly. NOTE: Prepare before use and avoid light.
- 0.2% formic acid, 50% acetonitrile, 10 mL
- Prepare 10 mL of 50% acetonitrile in deionized water.
- Add 20 µL of formic acid and vortex briefly.
- 0.2% formic acid solution, 10 mL
- Add 20 µL of formic acid to 10 mL of deionized water and vortex briefly.
- 0.1% formic acid solution, 10 mL
- Add 10 µL of formic acid to 10 mL of deionized water; vortex briefly.
3. Protein extraction in Schirmer strip
- Cut and discard the front end beyond the 0 mm mark of the strip to remove potential contamination due to the contact of conjunctival tissues with clean stainless-steel scissors.
- Cut the strip in 1 mm intervals into a 1.5 mL microcentrifuge tube (Figure 3).
- Add 100 µL of lysis buffer to the microcentrifuge tube, and vortex at 1,000 rpm, room temperature for 1 h in a thermomixer.
- Centrifuge briefly and transfer the supernatant to a new 1.5 mL microcentrifuge tube.
- Move the strips into 200 µL pipette tips using clean forceps. Place the tips into the same tube from step 3.4 with holder and centrifuge at room temperature at 4,000 × g for 1 min further for maximum recovery of the remaining sample on the strip.
- Measure protein concentration using bicinchoninic acid (BCA) or compatible protein assay.
4. Suspension-trapping sample preparation
- Normalize samples to 50 µg of protein based on the protein assay result.
- Add 200 mM DTT in a ratio of 1:10 (v/v, DTT: sample) to a final concentration of 20 mM DTT and incubate at 95 °C for 10 min.
- Cool down the protein solution to room temperature.
- Add 400 mM IAA in a ratio of 1:10 (v/v, IAA: sample) to a final concentration of 40 mM IAA and incubate in the dark at room temperature for 10 min.
- Add 12% aqueous phosphoric acid in a ratio of 1:10 (v/v, acid: sample) to a final concentration of 1.2% phosphoric acid and vortex briefly.
- Add protein binding buffer in a ratio of 1:6 (v/v, sample: buffer) and vortex briefly.
NOTE: In this step, colloidal protein particulate should be formed, and the solution will appear translucent if the protein amount is sufficient. - Uncap the Suspension Trapping Micro Spin Column and assemble the spin column onto a 2 mL microcentrifuge tube to collect flowthrough.
- Add up to 200 µL of acidified protein lysate mixture into the column.
- Centrifuge the column at 4,000 × g for 20 s. Protein particulates were trapped; repeat until the samples are loaded to the column.
- Add 150 µL of protein binding buffer and centrifuge at 4,000 × g for 20 s to wash the suspended protein. Repeat 3x.
- Assemble the Suspension Trapping Micro Column onto a new 1.5 mL microcentrifuge tube.
- Add 20 µL of 50mM TEAB digestion buffer containing protease at a ratio of 1:25 (w/w, trypsin: protein) onto the filter inside the spin column, avoiding bubbles and air gap between the filter and digestion solution.
- Cap the Suspension Trapping Micro Spin Column to prevent evaporation. Incubate at 47 °C for 1 h in a thermomixer; do not shake or vortex during digestion.
- Elute peptides with 40 µL of 50 mM TEAB by centrifugal force at 4,000 × g for 20 s.
- Add 40 µL of 0.2% FA and centrifuge at 4,000 × g for 20 s.
- Add 35 µL of 0.2% FA, 50% acetonitrile, and centrifuge at 4,000 × g for 20 s.
- Pool three eluates and dry with a vacuum centrifuge.
- Store at -80 °C ultra-low temperature freezer until further analysis.NOTE: The protocol can be paused here.
5. Reconstitute peptides for mass spectrometry analysis
- Reconstitute the sample with 12 µL of 0.1% FA, vortex, and centrifuge briefly.
- Measure peptide concentration with a Peptide Assay Kit.
- Normalize peptide concentration to 0.5 µg/µL in 0.1% FA.
- Transfer the peptide mixture to an autosampler vial and ready for mass spectrometry analysis.
6. Sample Acquisition by Liquid Chromatography-Tandem Mass Spectrometry
- Load 3 µg of the peptide with isocratic loading buffer (0.1% FA, 5% ACN) to a trap-column (C18, 5 µm, 100 µm, 20 mm) at a flow rate of 2 µL/min for 15 min.
- Fractionate the peptides using a reversed-phase nano analytical column (C18, 5 µm, 100 µm, 300 mm) at a flow rate of 350 nL/min in a 155 min separation gradient. Use mobile phase A comprising a mixture of 0.1% formic acid (v/v), 5% ACN (v/v) in water and mobile phase B containing 0.1% FA (v/v), 98% ACN (v/v) in water. Use the following gradient: 0-0.5 min: 5% B, 0.5-90 min: 10% B, 90-120 min: 20% B, 120-130 min: 28% B, 130-135 min: 45% B, 135-141 min: 80% B, 141-155 min: 5% B.
- Perform data-dependent acquisition (DDA) with a TOF-MS scan range between 350 to 1800 m/z with an accumulation time of 250 ms and a dynamic target exclusion time of 18 s. Then, carry out an MS/MS scan at 100 to 1,800 m/z in high-sensitivity mode with an accumulation time of 50 ms for the top 50 precursors per cycle with a threshold of 125 cps for MS/MS signal with a charge state from +2 to +4.
- Analyze raw data with the referenced software or other compatible engines with a reference database in FASTA from the publicly accessible UniProt database.
Representative Results
This protocol allows tear sample collection with Schirmer Strips that are stored dry at room temperature before subsequent sample preparation for mass spectrometry analysis. Filter-aided sample preparation (FASP) workflow with suspension-trapping micro column enabled fast sample preparation in hours, compared to commonly adopted in-solution digestion procedures in days that require overnight incubation. The peptide recovery yield was significantly higher (p < 0.001) than the standard in-solution digestion protocol and with good reproducibility at a coefficient of variation (%CV) < 7%. A pool of tears samples was repeated in six technical replicates with 74.2 ± 5.0% peptide recovery and 52.8 ± 1.6% peptide recovery in samples prepared with in-solution procedures. A pooled tear sample with a protein amount of 36.3 µg was spiked onto the Schirmer strip and extracted as previously described, the extraction efficiency of protein was 81% (29.5 ± 6.8 µg, mean ± SD).
Both workflows loaded 3 µg of peptides on the MS, and the DDA search yielded a total of 1,183 ± 118 proteins (5,757 ± 537 distinct peptides) and 874 ± 70 proteins (4,400 ± 328 peptides) at 1% FDR in the suspension trapping group and in-solution group, respectively. Analysis of gene ontology (GO) using the PANTHER classification system revealed very similar proteomes for both approaches, with binding, catalytic activity, and molecular function regulation being their main molecular functions (Figure 4).

Figure 1: Bend the top of the Schirmer strip at the 0 mm mark before application. Please click here to view a larger version of this figure.
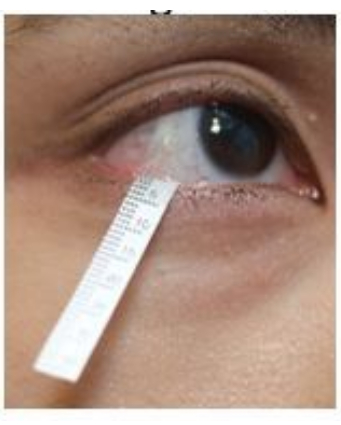
Figure 2: Position of the Schirmer strip during tear collection. Please click here to view a larger version of this figure.
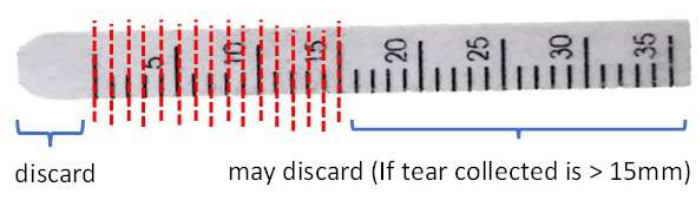
Figure 3: Illustration of Schirmer strip handling before protein extraction. Please click here to view a larger version of this figure.
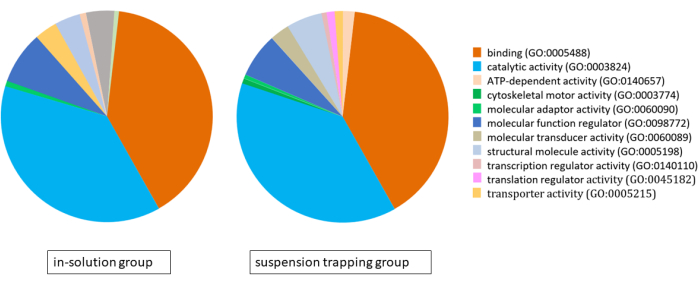
Figure 4: Pie chart illustrating the gene ontology analysis for the proteomes identified using the in-solution workflow and suspension trapping workflow, using the PANTHER classification system. Please click here to view a larger version of this figure.
Discussion
To achieve accurate results using this method, power-free disposable gloves should be worn in all procedures from tear sample collection to avoid sample contamination. It is important to avoid bubbles and air gaps between the filter and sample solution in each step by utilizing micro-spinning columns. If the sample volume is larger than the capacity of the columns, it is recommended to repeat the process. This protocol has proven to be more efficient than the traditional in-solution protocol in terms of preparation time, protein recovery, and total protein identification. This is largely because the samples undergo most of the required procedures on the same column, unlike the in-solution method which involves multiple transferring steps such as acetone precipitation, digestion, and cleanup (desalting), which increases the likelihood of variations in the resulting data.
In addition to the tear samples collected with the microcapillary method16,17, this FASP workflow with suspension-trapping micro column provides an alternative sample preparation method that allows fast and robust tear sample preparation collected using Schirmer strips, with minimal material preparation and user-friendly steps to follow. This allows reproducible tear sample preparation for large cohort studies across multiple ocular diseases or conditions with improved peptide recovery and protein identification by MS over in-solution procedures. This reliable process can be utilized regularly in the preparation of tear biomarker samples for research and other clinical purposes. Most importantly, it requires minimal staff training for on-site collection and negates the need for sample storage in a freezer. Samples are dried onsite to minimize protein autolysis and degradation. Hence, this allows convenient shipment by mail to facilitate downstream analysis, as opposed to using micro-capillary tubes.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
This work was supported by the InnoHK initiative and the Hong Kong Special Administrative Region Government; Research Centre for SHARP Vision; and the Research Centre for Chinese Medicine Innovation (RCMI) at The Hong Kong Polytechnic University.
Materials
| 9 mm Plastic Screw Thread Vials | Thermo Scientific | C4000-11 | |
| Acetonitrile, LCMS Grade | Anaqua | AC-1026 | |
| Centrifuge MiniSpin plus | Eppendorf | 5453000097 | |
| DL-dithiothreitol (DTT), BioUltra | Sigma-Aldrich | 43815 | |
| Eppendorf Safe-Lock Tubes, 1.5 mL | Eppendorf | 30120086 | |
| Formic acid, ACS reagent, ≥96% | Sigma-Aldrich | 695076 | |
| Frame Heater OPTIMONSUN Electronic | Breitfeld & Schliekert GmbH | 1203166 | |
| Iodoacetamide (IAA), BioUltra | Sigma-Aldrich | I1149 | |
| Methanol, HPLC Grade | Anaqua | MA-1292 | |
| Nunc Biobanking and Cell Culture Cryogenic Tubes, 2 mL | Thermo Fisher Scientific | 368632 | |
| Phosphoric acid, 85 wt.% in H2O | Sigma-Aldrich | 345245 | |
| Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher Scientific | 23275 | |
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo Fisher Scientific | A53225 | |
| Quadrupole Time-of-Flight Mass Spectrometry | Sciex | TripleTOF 6600 | |
| Schirmer Ophthalmic Strips | Entod Research Cell UK Ltd | I-DEW Tearstrips | |
| S-Trap Micro Column | Protifi | C02-micro-80 | |
| SureSTART 9 mm Screw Caps | Thermo Scientific | CHSC9-40 | |
| Triethylammonium bicarbonate (TEAB), 1 M | Sigma-Aldrich | 18597 | |
| Ultra-performance Liquid Chromatography | Eksigent | NanoLC 400 | |
| UltraPure Sodium dodecyl sulfate (SDS) | Thermo Fisher Scientific | 15525017 |
Referenzen
- Ma, J. Y. W., Sze, Y. H., Bian, J. F., Lam, T. C. Critical role of mass spectrometry proteomics in tear biomarker discovery for multifactorial ocular diseases (Review). International Journal of Molecular Medicine. 47 (5), 83 (2021).
- Tse, J. S. H., et al. Integrating clinical data and tear proteomics to assess efficacy, ocular surface status, and biomarker response after orthokeratology lens wear. Translational Vision Science & Technology. 10 (11), 18 (2021).
- Cheung, J. K., et al. Human tear proteome dataset in response to daily wear of water gradient contact lens using SWATH-MS approach. Data in Brief. 36, 107120 (2021).
- Tse, J. S., et al. Data on assessment of safety and tear proteome change in response to orthokeratology lens – Insight from integrating clinical data and next generation proteomics. Data in Brief. 29, 105186 (2020).
- Zhan, X., Li, J., Guo, Y., Golubnitschaja, O. Mass spectrometry analysis of human tear fluid biomarkers specific for ocular and systemic diseases in the context of 3P medicine. The EPMA Journal. 12 (4), 449-475 (2021).
- Ponzini, E., et al. Mass spectrometry-based tear proteomics for noninvasive biomarker discovery. Mass Spectrometry Reviews. 41 (5), 842-860 (2022).
- HaileMariam, M., et al. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. Journal of Proteome Research. 17 (9), 2917-2924 (2018).
- Ding, H., et al. Urine proteomics: evaluation of different sample preparation workflows for quantitative, reproducible, and improved depth of analysis. Journal of Proteome Research. 19 (4), 1857-1862 (2020).
- Ludwig, K. R., Schroll, M. M., Hummon, A. B. Comparison of in-solution, FASP, and S-Trap based digestion methods for bottom-up proteomic studies. Journal of Proteome Research. 17 (7), 2480-2490 (2018).
- Sze, Y. H., et al. High-pH reversed-phase fractionated neural retina proteome of normal growing C57BL/6 mouse. Scientific Data. 8 (1), 27 (2021).
- Marchione, D. M., et al. HYPERsol: High-quality data from archival FFPE tissue for clinical proteomics. Journal of Proteome Research. 19 (2), 973-983 (2020).
- Chhuon, C., et al. A sensitive S-Trap-based approach to the analysis of T cell lipid raft proteome. Journal of Lipid Research. 61 (11), 1512-1523 (2020).
- Hayoun, K., et al. Evaluation of sample preparation methods for fast proteotyping of microorganisms by tandem mass spectrometry. Frontiers in Microbiology. 10, 1985 (2019).
- Templeton, E. M., et al. Comparison of SPEED, S-Trap, and in-solution-based sample preparation methods for mass spectrometry in kidney tissue and plasma. International Journal of Molecular Sciences. 24 (7), 6290 (2023).
- Ding, Z., Wang, N., Ji, N., Chen, Z. S. Proteomics technologies for cancer liquid biopsies. Molecular Cancer. 21 (1), 53 (2022).
- Nättinen, J., Aapola, U., Jylhä, A., Vaajanen, A., Uusitalo, H. Comparison of capillary and Schirmer strip tear fluid sampling methods using SWATH-MS proteomics approach. Translational Vision Science & Technology. 9 (3), 16 (2020).
- Bachhuber, F., Huss, A., Senel, M., Tumani, H. Diagnostic biomarkers in tear fluid: from sampling to preanalytical processing. Scientific Reports. 11 (1), 10064 (2021).
