Using this protocol, single FDB muscle fibers were isolated and embedded in hydrogel. An overview of the muscle dissection procedure is shown in Figure 1. The FDB muscle is exposed with intact tendons and cut loose from the fascia. Maintaining the tendons of the muscles as fixation points minimizes potential damage to the muscle fibers during the isolation procedure. Excess connective tissue can be trimmed off to reduce debris and the growth of secondary cell types. Once the muscle has been excised and cleaned sufficiently, the muscle is enzymatically digested using collagenase, and single muscle fibers are released by trituration prior to embedding the isolated cells in hydrogels. The isolation of FDB muscle fibers provides relatively short muscle fibers, which have the advantage of being easily manipulated. Due to their size, FDB muscle fibers can be pipetted safely without tangle-induced damage and are easily embedded within a hydrogel. As single muscle fibers do not adhere to culture plates very well, embedding the fibers in hydrogel ensures that the fibers stay put during the cell culture and contractile measurements. Furthermore, the addition of a basement membrane matrix to the fibrin gel allows for interactions between the muscle fibers and the matrix, mimicking the native in vivo environment. Isolated single muscle fibers can be manipulated and maintained in culture for several days post isolation. In Figure 2, an example of isolated FDB fibers embedded in a hydrogel matrix is shown. The healthy fibers have visible sarcomeres and are stretched out straight (blue arrow), while the curved fibers are typically damaged or unviable (yellow arrow) and should be excluded from the measurements. Hypercontracted fibers appear as dark balled-up objects in the matrix (red arrow). If the isolation procedure was successful, the proportion of healthy fibers should be ~75%. A larger proportion of hypercontracted fibers usually indicates damage during the isolation procedure. The membrane of the muscle fibers can be damaged either by overdigestion of the muscle or damage to the fibers during trituration. Trituration damage mainly occurs if the muscle is underdigested and, thus, does not come apart easily.
The fiber functionality and viability were assessed through contractile measurements of the muscle fibers utilizing an optics-based, high-throughput contractile measurement system. The system outputs several parameters, such as the sarcomere length, percentage of sarcomere shortening, contractile velocity, and relaxation velocity. Using the contractile measurement system, sarcomere contraction can be measured per muscle fiber. Figure 3 shows examples of muscle fiber contractions measured using the measurement system. From a single contraction transient, the following parameters are obtained: sarcomere length at baseline, contraction duration, sarcomere length at maximal contraction, and relaxation duration (Figure 3A). These parameters are used to calculate the percentage of sarcomere shortening, contraction, and relaxation velocities. Average velocity values can also be calculated from the duration and absolute shortening values, if needed. A valid contraction measurement features a straight baseline, followed by a dip to the peak, and a return to the baseline (Figure 3A). Noise, unfocused sarcomeres, or aberrant movement of the fiber can influence the measurement transient (Figure 3B,C), and these measurements can be discarded manually or will be rejected by the analysis program. In this approach, the clear visualization of the sarcomeres is important for measuring the contractions; thus, anything that reduces the visibility of the sarcomeres may introduce noise. This could occur if there is movement of the sarcomere outside of the focal plane during contraction. Measurements in which a series of contractions differ in speed or depth should also be excluded from the dataset.
The contractile data of single muscle fibers obtained with this system can be used to compare different culture conditions. The effectiveness of the system is illustrated in Figure 4. Here, we measured the contractions of FDB muscle fibers in both 2D (laminin-coated culture plates) and 3D (fibrin hydrogel) culture formats. A higher percentage of usable measurements was achieved in 3D, as embedding the fibers in the gel prevented lateral movement and other movement artifacts from influencing the measurement (Figure 4A). Embedding the fibers had no significant effect on either the sarcomere shortening or maximum contraction speed values as compared to the 2D-cultured fibers (Figure 4B). To illustrate how different matrices influence the contraction of FDB muscle fibers, we also compared this fibrin hydrogel to a pure basement membrane matrix (4 mg/mL) (Figure 4C). The reduced contractility observed in the pure basement membrane matrix was likely due to the stiffness of the gel or increased cell-matrix interactions. Fibrin concentrations of up to 7 mg/mL have also been tested, with no significant effect on contractile speed and shortening (unpublished data). The usage of this fibrin-based hydrogel ensures minimal interference with contractile parameters.
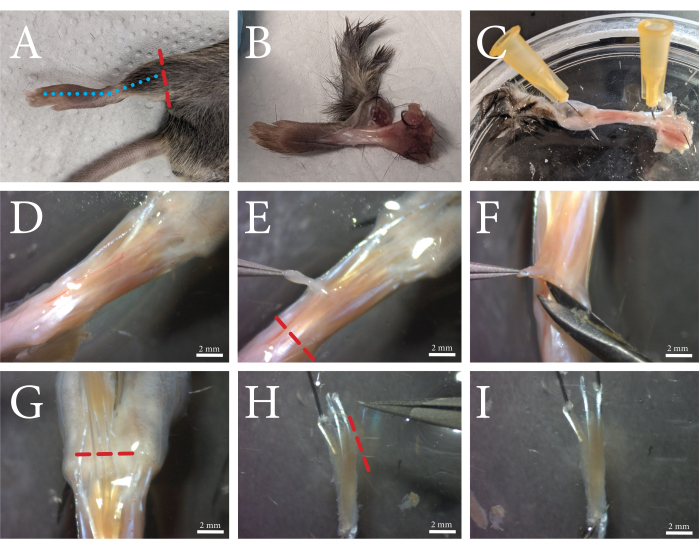
Figure 1: An overview of the FDB muscle dissection procedure. (A) The hind limb is cut above the ankle (red dashed line), and (B) the skin is removed by cutting along the top of the foot along the blue dashed line. (C) The foot is pinned through the ankle and the skin at the toes. (D) Microscopic view of the exposed muscle and its surrounding connective tissue. The fascia is visible as a white opaque layer through which the vasculature runs. (E) The fascia is removed from the muscle, and the tendon is cut along the red dashed line. (F) The FDB muscle is separated from the underlying tissue by lifting and cutting underneath and alongside the muscle. (G) When the tendons of the toes are clearly visible, the FDB is cut loose along the red dashed line. (H) The FDB is secured by the tendons, and the fourth lateral tendon and its fibers can be removed by cutting along the red dashed line. Excess fascia can now be trimmed off. (I) After cleaning, the FDB muscle is transferred to the collagenase solution. Scale bars = (D–I) 2 mm. Abbreviation: FDB = flexor digitorum brevis. Please click here to view a larger version of this figure.
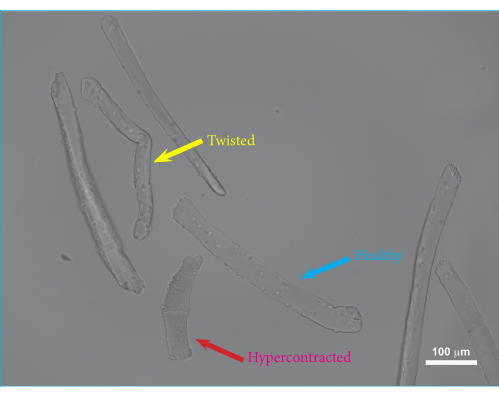
Figure 2: Microscopic image of FDB muscle fibers embedded in hydrogel after 24 h of culture. Blue arrow: Example of a viable FDB muscle fiber. Yellow arrow: Example of a twisted FDB muscle fiber. Twisted muscle fibers may have reduced viability and impaired contractions and, thus, should be excluded from the measurements. Red arrow: Example of a hypercontracted FDB muscle fiber. Excessive hypercontraction occurs when trituration is performed too vigorously or can be caused by collagenase overdigestion or the use of non-equilibrated culture medium. Scale bar = 100 µm. Abbreviation: FDB = flexor digitorum brevis. Please click here to view a larger version of this figure.
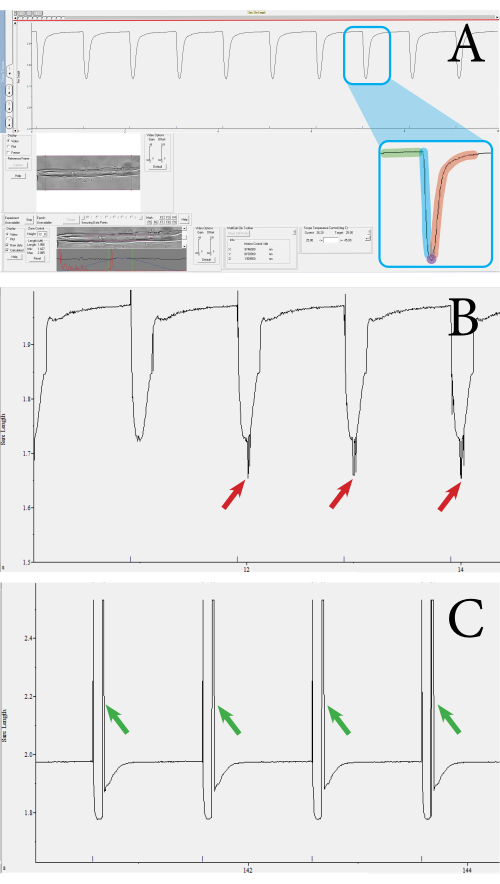
Figure 3: Example of fiber contraction transients measured using the optics-based, high-throughput contractile system. (A) Example of a normal contraction transient. This transient is obtained from the sarcomeres enclosed by the purple square shown in the lower toolbar. The transient consists of the following components: sarcomere length at baseline (green), contraction duration (blue), sarcomere length at maximal contraction (purple), and relaxation duration (red). Parameters such as the velocity and percentage of contraction are calculated from these values. (B) Example of an inadequate contraction transient. These measurements occur when the sarcomere signal is not picked up due to noise (see red arrows). (C) Example of a contraction transient that has a movement artifact (see green arrows). Movement artifacts occur when the muscle fiber moves outside the focus during the contraction. Please click here to view a larger version of this figure.
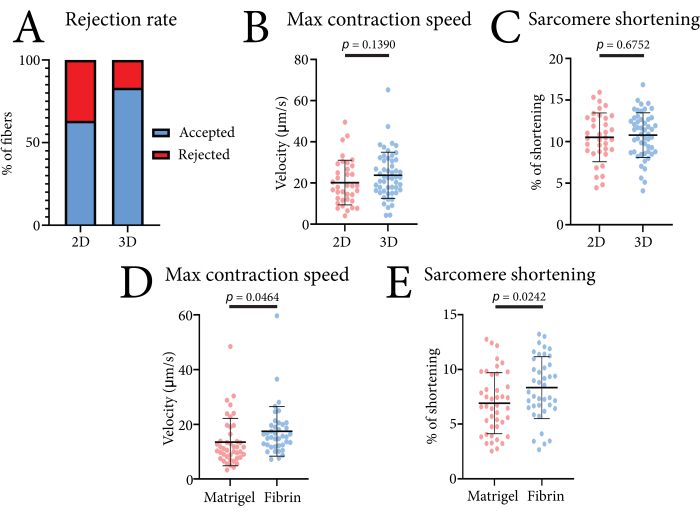
Figure 4: Representative data obtained using the optics-based high-throughput contractile system when comparing 2D versus 3D conditions and different hydrogels. (A) Percentage of accepted and rejected contraction measurements found by the data analysis program in 2D culture and in 3D culture based on 30 measurements. (B) Comparison of the maximum contraction speed in 2D-cultured and 3D-cultured muscle fibers isolated from three mice after 24 h of culture. (C) Comparison of the sarcomere shortening in 2D-cultured and 3D-cultured muscle fibers isolated from three mice after 24 h of culture. (D) Comparison of the maximum contraction speed of pure basement membrane matrix (Matrigel)-embedded and fibrin hydrogel-embedded muscle fibers isolated from three mice after 24 h of culture. (E) Comparison of the sarcomere shortening of pure basement membrane matrix (Matrigel)-embedded and fibrin hydrogel-embedded muscle fibers isolated from three mice after 24 h of culture. The data were analyzed using a Student's t-test and are shown as mean ± SD. Each data point is one muscle fiber. Please click here to view a larger version of this figure.
Table 1: Composition of the dissection medium used to dissect the muscle, the fibrin culture medium used to culture the isolated muscle fibers, and the muscle digestion medium used to enzymatically digest the isolated muscles. Please click here to download this Table.
Table 2: Composition of the cell mix used to cast the hydrogels and the matrix mix used to cast the hydrogels. Please click here to download this Table.
