Oxygen-Independent Assays to Measure Mitochondrial Function in Mammals
Summary
Here, we present a compilation of assays to directly measure mitochondrial function in mammalian cells independently of their ability to consume molecular oxygen.
Abstract
The flow of electrons in the mitochondrial electron transport chain (ETC) supports multifaceted biosynthetic, bioenergetic, and signaling functions in mammalian cells. As oxygen (O2) is the most ubiquitous terminal electron acceptor for the mammalian ETC, the O2 consumption rate is frequently used as a proxy for mitochondrial function. However, emerging research demonstrates that this parameter is not always indicative of mitochondrial function, as fumarate can be employed as an alternative electron acceptor to sustain mitochondrial functions in hypoxia. This article compiles a series of protocols that allow researchers to measure mitochondrial function independently of the O2 consumption rate. These assays are particularly useful when studying mitochondrial function in hypoxic environments. Specifically, we describe methods to measure mitochondrial ATP production, de novo pyrimidine biosynthesis, NADH oxidation by complex I, and superoxide production. In combination with classical respirometry experiments, these orthogonal and economical assays will provide researchers with a more comprehensive assessment of mitochondrial function in their system of interest.
Introduction
Mitochondrial function is a critical metric of cellular health, as it sustains key biosynthetic, bioenergetic, and signaling functions in mammalian cells1. The vast majority of mitochondrial functions require electron flow through the electron transport chain (ETC), and disruptions in electron flow in the ETC cause severe mitochondrial disease2. The ETC is comprised of a series of reduction and oxidation (redox) reactions that are embedded in the inner mitochondrial membrane, and these electron transfer reactions release free energy that can be harnessed to support ATP synthesis, physiological processes such as thermogenesis, biosynthetic pathways such as de novo pyrimidine biosynthesis, and the balance of the redox status of co-factors such as NADH. ETC complex I and III produce reactive oxygen species (ROS)3,4,5, which, in turn, regulate signaling key pathways such as HIF, PI3K, NRF2, NFκB, and MAPK6. Consequently, metrics of electron flow in the ETC are classically used as a proxy for mitochondrial function in mammalian cells.
Respirometry experiments are frequently employed to measure mitochondrial function in mammalian cells. Since O2 is the most ubiquitous terminal electron acceptor for the mammalian ETC, its reduction is used as a proxy for mitochondrial function. However, emerging evidence demonstrates that mammalian mitochondria can employ fumarate as an electron acceptor to sustain mitochondrial functions that depend on the ETC, including de novo pyrimidine biosynthesis7, NADH oxidation7, and the detoxification of hydrogen sulfide8. Thus, in certain contexts, especially in hypoxic environments, measurements of the O2 consumption rate (OCR) do not provide a precise or accurate indication of mitochondrial function.
Here, we outline a series of assays that can be employed to measure mitochondrial function independently of the OCR. We provide assays to directly measure complex I-mediated NADH oxidation, dihydroorotate dehydrogenase-mediated de novo pyrimidine biosynthesis, complex V-dependent ATP synthesis, the net directionality of the succinate dehydrogenase (SDH) complex, and mitochondrial-derived ROS. These assays are meant to be performed on cultured mammalian cells, although many can be adapted to study mitochondrial functions in vivo. Notably, the assays described in this protocol are more direct measurements of mitochondrial functions than the OCR. Moreover, they enable the measurement of mitochondrial function in hypoxia, a context in which the OCR is not an indicative measurement. Taken together, these assays, in combination with classical respirometry experiments, will provide researchers with a more comprehensive assessment of mitochondrial function in mammalian cells.
Protocol
1. Proliferation assays to measure the activity of complex I, dihydroorotate dehydrogenase (DHODH), and complex V activities
- Seeding cells for proliferation assays
NOTE: This protocol uses the human osteosarcoma cell line 143B purchased commercially from ATCC. This cell line was used under the guidelines of our approved Institutional Biosafety Committee (IBC) protocol.- Remove the plates from the tissue culture incubator. Aspirate the medium from the plates, and wash with 1x phosphate-buffered saline (PBS) to remove any leftover medium. Aspirate the PBS, and cover the dish with 0.05%-0.25% trypsin to lift the cells from the bottom of the plate.
- Wait 3-5 min for the trypsin to release the cells from the plate, and then quench the trypsin with 10 mL of the desired growth medium containing 10% fetal bovine serum (FBS).
- Collect the cells in a conical tube, and centrifuge at 1,000 × g for 5 min to pellet the cells.
- Aspirate the medium from the tube without disturbing the pellet. Resuspend the pellet with complete medium.
- Count the cells, and quantify the volume needed to seed between 10,000 and 25,000 cells in a 6-well plate.
NOTE: Each cell line will need to be optimized for the number of cells to be seeded. The ideal confluence achieved is ~10% at the beginning of the experiment and ~80% at the end of the experiment. - Pipette the cells into a 6-well plate, and add 2 mL of complete medium to the wells. Let sit for 24 h before changing to the experimental medium conditions.
NOTE: Seed at least three replicates per condition and enough wells to test the untreated control condition, inhibitor-treated control condition, untreated experimental condition, and inhibitor-treated experimental condition.
- Medium change to assess complex V activity by proliferation
NOTE: Cells proliferating in a medium with galactose are dependent on complex V activity for ATP synthesis9,10. Unlike glucose, which yields net two ATP from glycolysis, galactose yields none, forcing the cells to be dependent on complex V for ATP synthesis (Figure 1). The complex V inhibitor oligomycin is used as a control.- Make 10 mM glucose-containing medium (see Table 1).
- Make 10 mM galactose-containing medium (see Table 1).
- Change the medium in each well to either glucose-containing DMEM or galactose-containing DMEM. Add 5 µM oligomycin (the complex V inhibitor) to the relevant wells and the same volume of DMSO to the untreated wells. The oligomycin stock is 10 mM resuspended in DMSO. Put the plate back into the tissue culture incubator for 2 days.
- Medium change to assess complex I activity by proliferation
NOTE: Cells proliferating in a pyruvate-free medium are more reliant on complex I activity11,12. Without pyruvate, the cultured cells require complex I to facilitate the majority of NADH reoxidation back to NAD+ (Figure 2). The complex I inhibitor rotenone is used as a control.- Make pyruvate-free DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin.
- Make a 1 M solution of pyruvate (see Table 1).
- Change the medium in each well to either pyruvate-free medium or pyruvate-containing medium. Add 2 µM rotenone (the complex I inhibitor) to the treated wells and the same volume of DMSO to the untreated wells. The rotenone stock is 25 mM resuspended in DMSO. Put the plate back into the tissue culture incubator for 2 days.
- Medium change to assess DHODH activity by proliferation
NOTE: Cells proliferating in a uridine-free media require dihydroorotate dehydrogenase (DHODH) activity11,12,13. In the absence of exogenous uridine, the cultured cells synthesize pyrimidines through the de novo pathway. The DHODH inhibitor brequinar is used as a control.- Make uridine-free DMEM supplemented with 10% FBS and 1% penicillin-streptomycin.
- Make a 10 mg/mL uridine stock solution, and then prepare a 100 µg/mL uridine medium (see Table 1).
- Change the medium in each well to uridine-free medium or uridine-containing medium. Add 5 µM brequinar (the DHODH inhibitor) to the treated wells and the same volume of DMSO to the untreated wells. The brequinar stock is 10 mM resuspended in DMSO. Put the plate back into the tissue culture incubator for 2 days.
- Counting the cells for proliferation assays
- For all the experiments, replenish the medium every 2 days. If the medium becomes yellow in color, increase the frequency of the medium changes. Allow the cells to proliferate for up to 7 days, and stop the experiment if any of the wells begin to look overgrown. Wells are considered overgrown when the confluence exceeds 80%.
- Aspirate the medium, wash with 1x PBS, and cover the bottom of the well with 0.25% trypsin (500 µL for a 6-well dish).
- Wait 5 min until the cells have lifted off the dish. Check this under a microscope.
- Quench the trypsin with 1 mL of complete DMEM containing 10% FBS.
- Pipette up and down to break apart the cell clumps.
- Prepare Coulter counter cups by filling each with 10 mL of isotone buffer (one cup per well). Count the cells on a cell counter, and record the data. If the readout is cells per milliliter (cells/mL), multiply the recorded value by 1.5 to get the total number of cells per well.
NOTE: Other cell counting methods such as a hemocytometer will suffice if the lab does not have a Coulter counter.
2. 13C4-Aspartate stable isotope tracing and LC-MS analysis to measure DHODH activity
- 13C4-aspartate stable isotope tracing in adherent cells
NOTE: DHODH activity can be directly monitored by measuring the incorporation of 13C4-aspartate into 13C3-UMP. Brequinar is used as a control for DHODH activity (Figure 3). The resulting levels of 13C3-UMP are a measure of DHODH activity.- Seed between 250,000 and 500,000 cells in a 6-well dish to achieve 75% confluence the next day.
- Prepare a stock solution of 250 mM 13C4-aspartate and 10 mM 13C4-aspartate medium (see Table 1).
- Change the medium in each well to medium containing 10 mM 13C4-aspartate. Incubate for the appropriate number of hours to achieve a steady state for labeling the cells of interest.
NOTE: The steady state is defined as the time frame in which the percent labeled metabolite plateaus over time14. For 143B osteosarcoma cells, 13C4-aspartate achieves a steady state by 8 h. It is best practice to determine this time frame prior to the experimentation by doing a time course experiment with the stable isotope of interest.
- Metabolite isolation from the adherent cells
- Prior to beginning, prepare a bucket of dry ice, and prepare 80% HPLC-grade MeOH in HPLC-grade water. Cool this buffer in a −80 °C freezer overnight or place it directly on dry ice.
- Taking one plate out at a time from the incubator, aspirate the medium from the wells, and wash 2x with 1x PBS. Remove all the residual PBS from the wells before moving on to the next step.
NOTE: Make sure to tilt the plate during the aspiration and pipette against the wall of the dish to prevent the disruption of the adherent cells. - Place the plate on dry ice, and immediately add 800 µL of 80% LCMS-grade MeOH in 20% LCMS-grade water to each well.
- Incubate the plate for at least 15 min in a −80 °C freezer to facilitate cell lysis.
NOTE: At this point, the next plate can be taken out of the incubator and steps 2.2.1-2.2.4 repeated. Continue this until all the plates are incubating in the −80 °C freezer. - Taking one plate out at time from the freezer, scrape each well on dry ice using a cell lifter, and transfer the lysate to a 1.5 mL microcentrifuge tube. Keep the tube on dry ice until the next step.
- Vortex all the tubes for 10 min at 4 °C, and then centrifuge at 4 °C for 10 min at maximum speed (at least 17,000 × g).
- Transfer the supernatant to a 1.5 mL microcentrifuge tube, and dry down in a 4 °C vacuum concentrator equipped with a −105 °C cold trap on a high vacuum setting for approximately 6 h or until the samples have evaporated. Once the lysate is dried down, store the metabolite pellets at −80 °C until ready to prepare them for liquid chromatography paired with mass spectrometry (LCMS).
- Liquid chromatography-mass spectrometry (LC-MS) measurement of polar metabolites
NOTE: Any chromatographic and mass spectrometry workflow that allows for the detection of aspartate, the intermediates in de novo pyrimidine biosynthesis, and UMP will suffice14.- Prepare the samples for LCMS on ice by adding 100 µL of HPLC grade water to the dried pellets and vortex for 10 min at 4 °C.
- Centrifuge the samples at 4 °C for 10 min at maximum speed (at least 17,000 × g) and move 25 µL into each LC-MS vial.
- Inject 2 µL of lysate into the LC-MS system. A commonly used methodology is given below:
- Prepare mobile phase A comprising 20 mM ammonium carbonate (LC-MS grade) and 0.1% ammonium hydroxide (LC-MS Grade) dissolved in LC-MS grade water.
- Choose 100% acetonitrile (LC-MS Grade) as mobile phase B.
- For the chromatography, choose a 5 µm, 150 mm x 2.1 mm analytical column equipped with a 2.1 mm x 20 mm guard column for hydrophilic compounds (see the Table of Materials). Set the column oven to 25 °C.
- Use the following liquid chromatography settings: a constant flow rate of 0.15 mL/min; a linear gradient from 80% to 20% Mobile Phase B for 20 min, followed by a linear gradient from 20% to 80% Mobile Phase B for 0.5 min, followed by a hold at 80% Mobile Phase B for 7.5 min.
- Choose the following mass spectrometer settings: a full scan between m/z 70 Da and 1,000 Da; a resolution of 70,000; an AGC target of 1 × 106; and a maximum injection time of 20 ms. Operate the source in polarity switching mode. Set the spray voltage at 3.0 kV, the heated capillary at 275 °C, the HESI probe at 350 °C, the sheath gas flow at 40 units, the auxiliary gas flow at 15 units, and the sweep gas flow at 1 unit.
- Perform the data analysis using any software that interfaces with the LCMS workflow.
NOTE: The software used with the above workflow are XCalibur (Thermo) and TraceFinder (Thermo). Key considerations for data analysis are outlined below:- Expected retention times: Determine the retention time of each metabolite by running the standards for each metabolite of interest using the chromatographic method prior to the experiment.
NOTE: The retention times of UMP and its isotopologues, including 13C3-UMP, are the exact same. - Expected M/Z for metabolites: If a metabolite ionizes in the negative ion mode (such as UMP), calculate the expected exact mass using the molecular formula of UMP minus a proton [C9H14N2O9P]. For metabolites that ionize in positive ion mode, calculate the exact mass by using the molecular formula plus a proton.
- Mass accuracy: When using an orbitrap mass spectrometer, the metabolite of interest and its isotopologues are expected to have a mass accuracy ±5 mmu. Use software to calculate this, accounting for the expected M/Z and the actual detected M/Z.
- Natural abundance correction: All tracing data are expected to undergo natural abundance correction to account for the ~1% 13C and ~0.5% 15N-isotopes in nature15.
- Expected retention times: Determine the retention time of each metabolite by running the standards for each metabolite of interest using the chromatographic method prior to the experiment.
3. 13C5-glutamine stable isotope tracing to measure SDH activity
- 13C5-glutamine stable isotope tracing in adherent cells
NOTE: SDH activity can be directly monitored by measuring the interconversion of certain isotopologues of fumarate and succinate upon 13C5-glutamine tracing7,16,17. The complex III inhibitor antimycin is used as a control to alter the net directionality of the SDH complex (Figure 4).- Seed between 250,000 and 500,000 cells in a 6-well dish to achieve 75% confluence the next day.
- Prepare a stock of 50 mM 13C5-glutamine (see Table 1).
- Prepare tracing medium containing 2 mM 13C5-glutamine (see Table 1).
- Change the medium in each well to medium containing 2 mM 13C5-glutamine. Incubate for the appropriate number of hours to achieve a steady state of labeling the cells of interest (see the note in step 2.1.3).
- Isolate the metabolites following the protocol in step 2.2.
- Analyze the samples on LC-MS following the workflow described in step 2.3.
- Analyzing the SDH activity based on labeling patterns
NOTE: The succinate oxidation activity of the SDH complex can be monitored by assessing the ratio of 13C4-fumarate to 13C4-succinate upon 13C5-glutamine tracing. The fumarate reduction activity of the SDH complex can be monitored by assessing the ratio of 13C3-succinate to 13C3-fumarate upon 13C5-glutamine tracing.- Calculate the percent labeling of 13C3-fumarate, 13C4-fumarate, 13C3-succinate, and 13C4-succinate. For example, to determine the percentage of 13C3-fumarate, sum up all the integrated peak areas for the fumarate isotopologues (unlabeled fumarate, 13C1-fumarate, 13C2-fumarate, 13C3-fumarate, and 13C4-fumarate), and divide the peak area for 13C3-fumarate by the total isotopologue area. Multiply by 100.
- To calculate the succinate oxidation, divide the percentage 13C4-fumarate by the percentage 13C4-succinate.
- To calculate the fumarate reduction, divide the percentage 13C3-succinate by the percentage 13C3-fumarate.
4. Direct complex I activity assay
NOTE: DCPIP is an artificial electron acceptor; it changes to its reduced form when accepting electrons from ubiquinol. In this assay, ubiquinone is reduced to ubiquinol via the complex I-mediated oxidation of NADH to NAD+. Thus, measuring the turnover of oxidized DCPIP in this cell-free assay is a proxy for complex I activity7,18.
- Purifying and quantifying the mitochondria from cells
NOTE: Any mitochondrial purification protocol can be utilized for this assay19.- Expand the cells until 25-100 million cells are obtained. Aspirate the medium off the dishes, wash with 1x PBS, aspirate the PBS, and tryspinize the cells with 0.25% trypsin. Quench the trypsin with culture medium, and pellet the cells at 1,000 × g for 5 min.
- Wash the cell pellets 2x in 5 mL of 1x PBS, and repeat the centrifugation at 1,000 × g for 5 min to pellet the cells. Store the washed pellets in a −20 °C freezer until mitochondrial purification.
NOTE: Do not freeze-thaw the cell pellets more than once. - Prepare mitochondria isolation buffer (see Table 1).
NOTE: Sodium-containing reagents such as NaOH should not be used to prepare mitochondria isolation buffers, as sodium tarnishes the mitochondrial membrane potential20 and disrupts mitochondrial calcium homeostasis21. - Resuspend the cell pellets to 10 million cells per 1 mL of mitochondria isolation buffer. For example, resuspend 100 million pelleted cells in 10 mL of mitochondria isolation buffer.
NOTE: Make sure to disrupt the cell clumps without introducing bubbles into the buffer. - Move 2 mL of cell suspension into a precooled glass homogenizer with a working volume of 3-8 mL that is compatible with a Potter-Elvehjem PTFE pestle. Homogenize the cells with 10-20 strokes, monitoring the cells under a microscope to confirm cell lysis throughout the homogenization process. Increase the number of strokes if needed to ensure efficient cell lysis.
NOTE: If homogenizing the cells with the above method is not effective for lysing the cells, switch to a syringe lysis method22. - Transfer 2 mL of the cell lysate into a microcentrifuge tube on ice, and repeat the above steps until the entire cell suspension is homogenized.
- Pellet the nuclei and cell debris at 650 × g for 10 min in a 4 °C centrifuge.
- Move the supernatant to a new 2.0 mL tube, and repeat the above centrifugation at 650 × g for 10 min at 4 °C.
- Move the supernatant to a new 2.0 mL tube, and pellet the crude mitochondria at 7,000 × g for 10 min in a 4 °C centrifuge.
- Discard the supernatant, and resuspend the pellet in 1 mL of mitochondria isolation buffer. Move 50 µL into a new tube for protein quantification. Repeat the above centrifugation step on the two aliquots of the sample (the 50 µL sample and the remaining ~950 µL).
- Discard the supernatant, and store the pellets in a −80 °C freezer until ready to use.
- Add 200 µL of RIPA buffer to the pellets from the 50 µL aliquot to extract the protein, vortex for 10 min, and centrifuge at 21,000 × g to isolate the protein. Quantify the amount of protein in the 50 µL aliquot, and use this number to quantify the remaining protein in the 950 µL sample. For example, if the protein quantification for the 50 µL aliquot is 1 µg/µL, the total protein in the remaining mitochondrial pellet is 950 µg (1 µg/µL × 950 µL).
- Performing the complex I activity assay
NOTE: The 143B osteosarcoma cell line was utilized for this assay, but the protocol can be adapted for any cultured cells.- Resuspend purified mitochondria in the mitochondria resuspension buffer (see Table 1) to a final concentration of 5 mg protein/mL.
- Perform five freeze/thaw cycles in a −20 °C freezer to permeabilize the mitochondria.
- Make the complex I activity assay buffer (see Table 1).
- Mix 50 ug of the 5 mg/mL mitochondria stock with 90 µL of complex I activity buffer. Prepare five untreated replicates and five rotenone-treated (5 µM) replicates to control for complex I activity.
- Read the baseline absorbance at 600 nm in a plate reader set to 37 °C.
- Initiate the reaction by adding NADH to a final concentration of 2 mM, and track the absorbance (600 nm) over the span of 1 h, measuring at least every 2 min throughout. The decrease in absorbance is indicative of a reduction of DCPIP via complex I activity.
NOTE: The NADH should be added using a multichannel pipette to ensure that all the samples are initiated at the same time.
5. LC-MS-based assay to measure the superoxide levels
NOTE: The fluorescence properties of MitoSox Red can change independently of its reaction with superoxide23. This LC-MS-based assay directly measures the product from superoxide reacting with MitoSox Red. The following assay is slightly modified from Xiao et al.24. 2-Hydroxy-mitoethidium (2-OH MitoE2+) is the product of the superoxide reaction (Figure 5). The Caki1 cell line was utilized for this assay, but the protocol can be adapted for any cultured cells.
- Treating adherent cells with MitoSox Red
- Seed between 250,000 and 500,000 cells in a 6-well dish to achieve 75% confluence the next day. Be sure to seed a set of plates for the LC-MS assay and a duplicate set of plates for cell count normalization.
- Prepare complete DMEM containing 10% heat-inactivated FBS and 1% penicillin-streptomycin in Dulbecco's modified Eagle medium.
- Refresh the medium in all the wells for all the conditions with 2 mL of complete DMEM. Make sure to add in any drugs or treatments of interest at this point.
- Incubate the cells in a tissue culture incubator for 1 h.
- Prepare positive and negative controls. Prepare a 50 mM stock of tert-butyl hydroperoxide diluted in PBS and a 250 mM stock of N-acetyl cysteine (NAC) diluted in DMSO (see Table 1).
NOTE: Tertbutyl hydroperoxide (tBuOOH) is a potent reactive oxygen species that will act as a positive control and increases the amount of the 2-OH MitoE2+. NAC is a potent antioxidant that will act as a negative control and neutralize the superoxide production caused by tBuOOH. - Add 1 µL of tBuOOH to the positive control wells and 1 µL of tBuOOH + 100 µL of NAC to the negative control wells.
NOTE: Using a P2 pipette (0.1-2 µL range) is the optimal way to transfer 1 µL. - Incubate for 1 h in a tissue culture incubator.
- Prepare 1 mM MitoSox Red stock (see Table 1), and add 2 µL of 1 mM MitoSox Red to each well to achieve a final concentration of 1 µM. Incubate for 30 min in a tissue culture incubator.
- During this final incubation, count one of the duplicate plates to acquire the cell counts to normalize the final LC-MS data.
- Isolation of the oxidized Mitosox Red product from the adherent cells
- Get a bucket of dry ice, and place cool HPLC-grade isopropanol on the dry ice prior to beginning the isolation.
- Taking one plate out at a time, aspirate the medium from the wells, and wash 2x with 1 mL of 1x PBS. Aspirate all the residual PBS from the wells before moving on to the next step.
NOTE: Make sure to tilt the plate during the aspiration and pipette against the wall of the dish to prevent disruption of the adherent cells. - Place the plate on dry ice, and add 800 µL of HPLC-grade isopropanol to each well.
- Incubate the plate for at least 15 min in a −80 °C freezer to facilitate cell lysis.
NOTE: At this point, the next plate can be taken out of the incubator and steps 5.2.1-5.2.4 repeated. Continue this until all the plates are incubating in the −80 °C freezer. - Taking one plate out at a time from the freezer, scrape each well on dry ice using a cell lifter, and transfer the lysate to a 1.5 mL microcentrifuge tube. Keep the tube on dry ice until the next step.
- Vortex all the tubes for 10 min at 4 °C, and then centrifuge at 4 °C for 10 min at maximum speed (at least 17,000 × g).
- Transfer the supernatant to a 1.5 mL microcentrifuge tube, and dry down in a vacuum concentrator. Once the lysate is dried down, store the metabolite pellets at −80 °C until ready for LC-MS.
- LC-MS measurement of MitoSox Red and 2-hydroxy-mitoethidium (2-OH MitoE2+)
- Prepare the samples for LCMS on ice by adding 100 µL of a 3:3:1 mixture of HPLC-grade MeOH:chloroform:water. Vortex for 10 min at 4 °C.
- Move 25 µL into each LC-MS vial. Store the remaining sample in the −80 °C freezer.
- Prepare the following liquid chromatography buffers: i) buffer A: HPLC grade water with 0.1% formic acid; ii) buffer B: HPLC grade acetonitrile with 0.1% formic acid.
- Open the Thermo XCalibur Instrument Setup app to begin developing the method.
- Look for two boxes on the left sidebar of this window-the first box allows chromatography method development, and the second box allows mass spectrometry method development.
- Liquid chromatography method development:
- Set a flow rate of 0.25 mL/min, and start the method at 20% B, rising to 95% B over the span of 12 min. Over the next minute, drop buffer B back down to 20% B, and maintain that level for the next 2 min.
NOTE: The last 2 min of flow at 20% B is critical for the column pressure to drop back down to the initial pressure and to equilibrate the column with the appropriate buffer ratios. Not including this equilibration step will cause the retention times to shift for all the subsequently run samples.
- Set a flow rate of 0.25 mL/min, and start the method at 20% B, rising to 95% B over the span of 12 min. Over the next minute, drop buffer B back down to 20% B, and maintain that level for the next 2 min.
- Create a Tune File with the following parameters:
- Open the Tune App, and create a new tune file.
- Apply any overlapped settings from the Method Setup module (see step 5.3.9.3) to this tune file as well, including the scan range, resolution, polarity, AGC target, and maximum IT.
- Set the Sheath gas auf 30, the Aux gas auf 3, and the Sweep gas auf 3 as well. Set the Spray voltage auf 3 kV, the Capillary temp auf 300, and the S-lens RF level auf 60.
- Mass spectrometry method development:
- From the bottom left list of potential scans, drag and drop Full MS to the center of the window. Make sure to click on the Full MS square after the drop to adjust the settings. The total MS method duration is 13 min.
NOTE: The LC method is longer than the MS method because the final 2 min are for column reconditioning and not metabolite quantification. - On the right side of the module, under Properties of the method, make sure the Method duration and Run time are set to 13.00 min.
- Run this full scan in positive mode at a Resolution of 70,000 and an AGC target of 1 × 106. Set the Maximum IT auf 100 ms and the Scan range from 300 m/z auf 700 m/z.
- Under Tune Files toward the top of the module, observe that the tune file that was just created is linked to this method.
- From the bottom left list of potential scans, drag and drop Full MS to the center of the window. Make sure to click on the Full MS square after the drop to adjust the settings. The total MS method duration is 13 min.
- Run the method for MitoSox Red detection with 2 µL sample injections.
Representative Results
The activities of DHODH, complex I, and complex V can all be assessed using proliferation assays. Upon deprivation of uridine from the culture medium, the cells become more dependent on the de novo pathway for pyrimidine biosynthesis. Thus, when cells were challenged to proliferate in a uridine-free medium, they were more sensitive to the inhibition of DHODH activity by brequinar than cells cultured in a medium containing uridine (Figure 6A). Similarly, the deprivation of pyruvate from the culture medium renders the cells more dependent on complex I activity for proliferation. Thus, when cells were challenged to proliferate in a pyruvate-free medium, they were more sensitive to the inhibition of complex I activity by rotenone than cells cultured in a pyruvate-containing medium (Figure 6B). Complex V activity can be assessed by challenging cells to proliferate in a medium containing galactose instead of glucose. As galactose yields net zero ATP in glycolysis, cells growing in this fuel are more reliant on mitochondrial ATP synthesis via complex V activity. Thus, cells proliferating in a galactose-containing medium were more sensitive to complex V inhibition by oligomycin than cells proliferating in a glucose-containing medium (Figure 6C).
SDH activity can be measured using 13C5-glutamine tracing and by monitoring its incorporation into fumarate and succinate isotopologues. In vehicle-treated conditions, the SDH complex favored the forward activity, and the incorporation of 13C4-succinate into 13C4-fumarate was higher than the incorporation of 13C3-fumarate into 13C3-succinate (Figure 7). In antimycin-treated conditions, the SDH complex favored the reverse activity, and the incorporation of 13C3-fumarate into 13C3-succinate was greater than the incorporation of 13C4-succinate into 13C4-fumarate (Figure 7).
Superoxide production inside the mitochondria can be measured using the fluorescent reporter MitoSox, which generates 2-hydroxy-mitoethidium upon reaction with superoxide. In this study, cells treated with MitoSox in the presence of tertbutyl hydrogen peroxide had higher levels of 2-hydroxy-mitoethidium in a manner that was suppressed by the addition of NAC, an antioxidant that quenches cellular ROS (Figure 8).
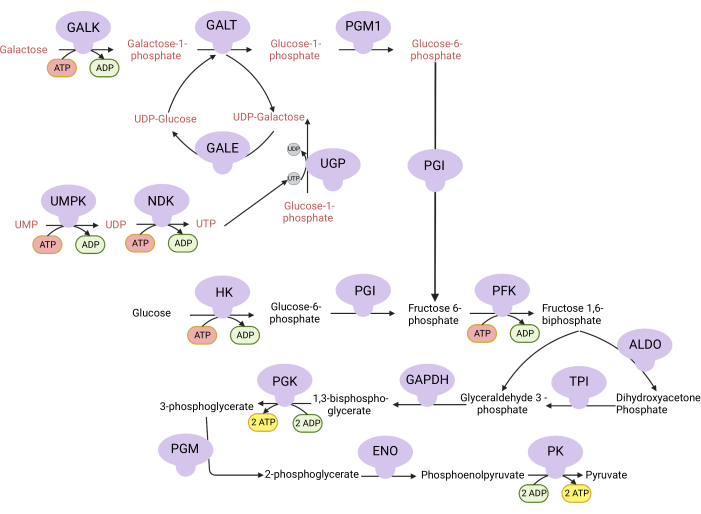
Figure 1: Mechanistic basis for the complex V proliferation assay. Oxidation of glucose and galactose via glycolysis. Glucose yields net two ATP from glycolysis, whereas galactose yields net zero ATP because UTP synthesis is required for UDP-galactose. Thus, cells grown in galactose are more dependent on mitochondrial ATP synthesis due to a lack of ATP produced from glycolysis. Abbreviations: GALK = galactokinase; GALT = galactose-1-phosphate uridylyltransferase; PGM1 = phosphoglucomutase 1; GPI = glucose-6-phosphate isomerase, UGP = UDP-glucose pyrophosphorylase; GALE = UDP-galactose-4-epimerase; NDK = nucleotide diphosphate kinase; UMPK = uridine monophosphate kinase; HK = hexokinase; PFK = phosphofructokinase; ALDO = aldolase; TPI = triosephosphate isomerase; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; PGK = phosphoglycerate kinase; PGM = phosphoglucomutase; ENO = enolase; PK = pyruvate kinase. Please click here to view a larger version of this figure.
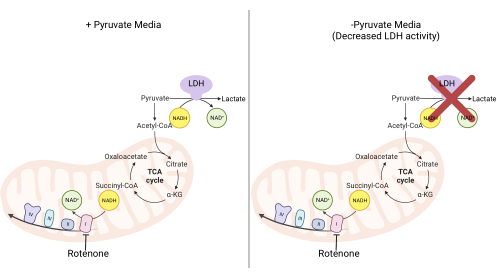
Figure 2: Mechanistic basis for the complex I proliferation assay. Schematic of the metabolic pathways that are altered upon the inhibition of complex I activity. In high-pyruvate media, complex I inhibition is bypassed via LDH-mediated NADH oxidation. In low-pyruvate media, this adaptation is less feasible, making cells more reliant on complex I activity to reoxidize NADH. Abbreviations: LDH = lactate dehydrogenase; TCA cycle = tricarboxylic acid cycle. Please click here to view a larger version of this figure.
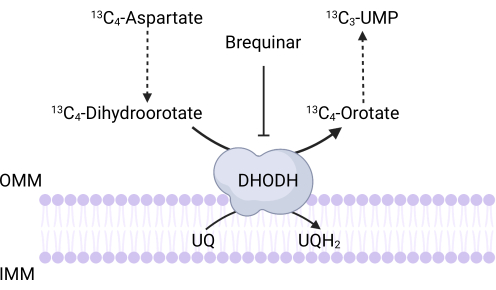
Figure 3: Schematic of the DHODH reaction upon 13C4-aspartate tracing. Brequinar inhibits dihydroorotate oxidation to orotate, thus preventing the downstream synthesis of UMP. 13C4-aspartate is incorporated into 13C3-UMP via DHODH activity. Abbreviations: OMM = outer mitochondrial membrane; IMM = inner mitochondrial membrane; DHODH = dihydroorotate dehydrogenase. Please click here to view a larger version of this figure.
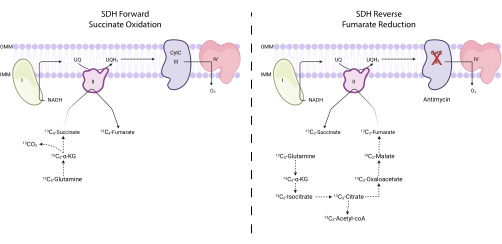
Figure 4: Measuring forward and reverse complex II activity via 13C5-glutamine tracing. To measure forward complex II activity (left), the incorporation of 13C5-glutamine into 13C4-succinate and 13C4-fumarate is monitored. To measure reverse complex II activity (right), the incorporation of 13C5-glutamine into 13C3-succinate and 13C3-fumarate is monitored. Abbreviations: SDH = succinate dehydrogenase; CytC = cytochrome C. Please click here to view a larger version of this figure.
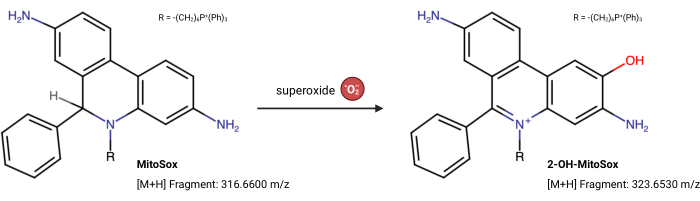
Figure 5: MitoSox Red reaction with superoxide. The reaction of MitoSox with mitochondrial superoxides to form 2-OH-MitoE2+. Please click here to view a larger version of this figure.
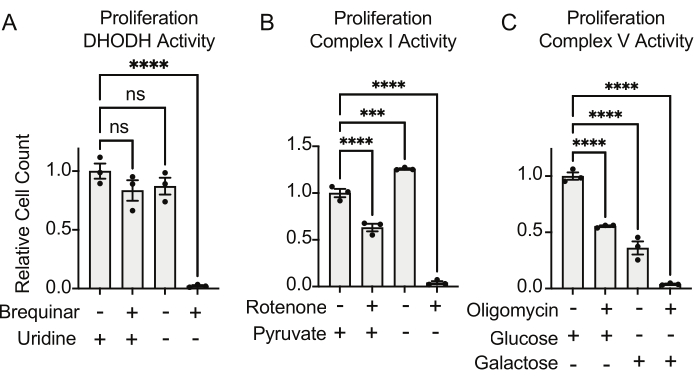
Figure 6: Proliferation-based assays to measure mitochondrial function (A) Proliferation of 143B osteosarcoma cells treated with 5 uM brequinar, a DHODH inhibitor, in medium with ±100 ug/mL uridine. Data are mean ± SEM; N = 3 per condition. (B) Proliferation of 143B osteosarcoma cells treated with the 2 uM rotenone, a complex I inhibitor, in medium with ±5 mM pyruvate. Data are mean ± SEM; N = 3 per condition. (C) Proliferation of 143B osteosarcoma cells treated with the 5 uM oligomycin, a complex V inhibitor, in medium with either 10 mM glucose or 10 mM galactose as the sole central carbon source. Data are mean ± SEM; N = 3 per condition. * indicates p < 0.05 using a one-way ANOVA test in Graphpad Prism. Please click here to view a larger version of this figure.
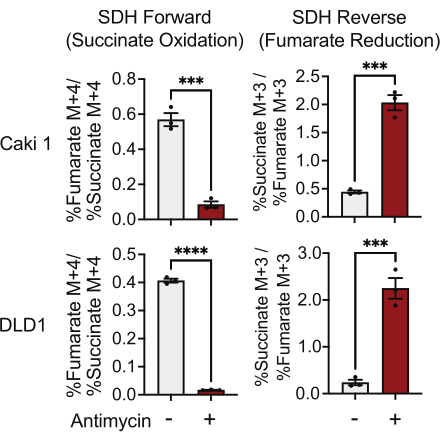
Figure 7: 13C5-glutamine tracing to measure complex II activity. Succinate oxidation and fumarate reduction (SDH reverse) in DMSO and 500 nM antimycin A-treated Caki1 and DLD1 cells. Data represent mean ± SEM; N = 3 per condition. *** indicates p < 0.05 using an unpaired t-test in GraphPad Prism. Abbreviations: SDH = succinate oxidation; SDH reverse = fumarate reduction. Please click here to view a larger version of this figure.
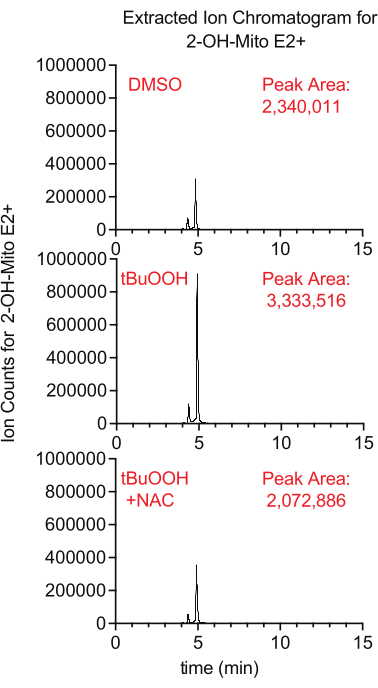
Figure 8: LCMS-based MitoSox assay to detect superoxide. Extracted ion chromatogram of 2-OH-Mito E2+ isolated from Caki1 cells treated with MitoSox for 30 min in the presence of tBuOOH ± NAC. Abbreviations: LCMS = liquid chromatography-mass spectrometry; NAC = N-acetyl cysteine. Please click here to view a larger version of this figure.
Table 1: Composition of reagents, buffers, and media used in this protocol. Please click here to download this Table.
Discussion
As emerging research demonstrates that mammalian mitochondria can function without consuming molecular oxygen, it is of the utmost importance for researchers to employ orthogonal assays, beyond OCR measurements, to accurately quantify mitochondrial function. Here, we compiled a series of assays that can be used to directly assess the activities of complex I, complex II, complex V, and DHODH by measuring the mitochondrial NAD+/NADH balance, the utilization of adaptive terminal electron acceptors, the production of ATP, de novo pyrimidine biosynthesis, and mitochondrial-derived ROS. Notably, these assays more directly measure mitochondrial function than OCR measurements. Furthermore, these assays provide researchers with tractable ways to quantify mitochondrial function during hypoxia, for which OCR measurements are largely irrelevant due to fumarate being used as the favored terminal electron acceptor. Finally, the proliferation-based methods described here are more cost-effective than classical respirometry experiments, thus providing a broadly accessible way to study mitochondrial function in mammalian systems.
There are key considerations when utilizing these assays to measure mitochondrial function in cultured cells. Regarding the proliferation assays, it is important to adjust the number of cells seeded for the doubling rate of each cell line. The cells should be seeded to at least 10% confluence and with enough space to allow for three to four doublings so that differences in proliferation can be quantified. Another consideration for each assay is the concentration of the small molecules used as controls for the activities of each ETC complex. As different cell lines may exhibit different sensitivities to these inhibitors, it is critical to test the dose of these small molecules to identify the optimal concentration.
A universal limitation of assays studying mitochondrial function in vitro, including OCR measurements and all the assays described here, is the metabolic composition of the culture medium. Standard cell culture medium tends to bias systems into superficially high levels of mitochondrial function. For example, supraphysiological glutamine levels increase its anaplerosis of the TCA cycle25, which fuels mitochondrial NADH synthesis and, consequently, increases oxidative phosphorylation. Similarly, the partial pressure of oxygen ranges between 3 mmHg and 100 mmHg (approximately 0.1%-13% O2) in mammalian tissues but is atmospheric (140 mmHg, approximately 21%) in vitro26,27. This excess O2 maximizes the mitochondrial respiratory capacity and superoxide production28. Recently, efforts have been made to design culture media to be more physiological29,30. Notably, culturing cells in human plasma-like media decreases mitochondrial respiration in some cancer cell lines30, mitochondrial ROS in T cells31, and mitochondrial adaptations to cancer therapeutics32. Thus, it is critical to be mindful of the composition of the culture media being used and understand how it may impact the mitochondrial function.
Another important and universal limitation in the interpretation of mitochondrial function is the potential for differences in the number of mitochondria. It is, therefore, critical to measure the mitochondrial content through either the quantification of mtDNA33, the measurement of mitochondrial mass with membrane potential-insensitive dyes34, or western blotting of mitochondrial markers. This is a critical control so that a decrease in the number of mitochondria is not mistaken for a decrease in mitochondrial function.
There are also specific limitations and troubleshooting that apply to the assays described here. First, given that differentiated cells do not proliferate, the proliferation-based assays will not be useful for assessing mitochondrial function in this context. A key limitation of the 13C4-aspartate tracing protocol to measure DHODH activity is that aspartate uptake in cells can be extremely inefficient35. To overcome this potential limitation, researchers can overexpress the aspartate transporter, SLC1A3, to facilitate 13C4-aspartate uptake35.
A limitation of the protocol using 13C5-glutamine tracing to measure SDH activity is that this assay requires cells to utilize the reductive carboxylation pathway to enrich the M+3 isotopologues in order to measure the reverse activity. Some cell lines are incapable of reductive carboxylation flux due to low ATP citrate lyase expression36, insufficient HIF stabilization37, or an α-KG:citrate ratio that is too low38. To overcome this limitation, one could utilize 13C4-aspartate tracing to measure the SDH forward and reverse activities7. In this assay, the SDH forward activity can be measured by the ratio of fumarate M+2:succinate M+2 and the reverse reaction by succinate M+4:fumarate M+4. Notably, this tracing circumvents most of the enzymes in the reductive carboxylation pathway.
A limitation of the complex I activity assay using DCPIP reduction as the readout is that the mitochondria are not structurally intact. The process of freeze-thawing the mitochondria to enable their NADH uptake for the assay can certainly tarnish the structural integrity of the mitochondrial membrane39. This assay should be performed in parallel with assays such as the complex I proliferation assay to ensure that the changes in complex I activity observed are also true with intact cells.
In future studies, some of these techniques can be adapted for measuring mitochondrial functions in vivo using model organisms such as mice and Caenorhabditis elegans. The current methods used to measure mitochondrial function in vivo are centered on the organismal-level OCR, specifically the respiratory exchange rate when using mouse models. A clear limitation of this method is that oxygen serves many biochemical and signaling functions beyond its role as a ubiquitous terminal electron acceptor in the mitochondrial ETC. For example, oxygen is "consumed" by the catalytic activity of enzymes in the dioxygenase family. Although these enzymes contribute to the cellular oxygen consumption rate, they do not participate in, regulate, or reflect mitochondrial function. Classical respirometry experiments in vitro typically control for "non-mitochondrial OCR", whereas organismal respiratory exchange ratio (RER) experiments cannot control for this, limiting the interpretation of RER as a metric for mitochondrial function in vivo. However, it is feasible to adapt the protocols to measure DHODH activity via 13C4-aspartate tracing, complex II activity via 13C5-glutamine tracing, complex I activity on mitochondria purified from tissues, and mitochondrial ROS using LC-MS friendly compounds such as MitoB in order to measure mitochondrial function in vivo. These direct assays to interrogate mitochondrial functions, in combination with classical respirometry experiments, provide researchers with a more comprehensive and accurate assessment of mitochondrial function in mammalian cells and tissues.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
The figures produced in this manuscript were created with BioRender.com. We are grateful to Amy Walker for providing feedback on this article. J.B.S. was supported by the Worcester Foundation for Biomedical Research Grant.
Materials
| 1.5 mL tube | Cell Treat | 667443 | |
| 2.0 mL tube | Cell Treat | 229446 | |
| 6-well plate | Cell Treat | 229106 | |
| 12-well plate | Cell Treat | 229112 | |
| 13C4-aspartate | Sigma-Aldrich | 604852 | |
| 13C5-Glutamine | Cambridge Isotope Laboratories | 285978-14-5 | |
| 15 mL centrifuge tube | Cell Treat | 667411 | |
| 50 mL centrifuge tube | Cell Treat | 667421 | |
| 150 mm tissue culture dish | Cell Treat | 229651 | |
| 1x Phosphate-buffered saline | Gibco | 10010049 | |
| 2,6-dichlorophenolindophenol | Honeywell | 33125 | |
| Ammonium Carbonate | Sigma-Aldrich | 37999 | |
| Antimycin | Sigma-Aldrich | A8674 | |
| Ascentis Express C18 | Sigma-Aldrich | 53825-U | |
| Bottle top filter 500 mL, 0.22 µm, PES 9 9 mm membrane diameter | Cell Treat | 229717 | |
| Bovine Serum Albumin | Sigma-Aldrich | A3294 | |
| Brequinar | Sigma-Aldrich | SML0113 | |
| Cell Lifter, Double End Flat and Narrow Blade | Cell Treat | 229305 | |
| CentriVap -105 Cold Trap | Labconco | 7385020 | |
| Complete Protease Inhibitor Tablets | Sigma-Aldrich | 4693116001 | |
| Coulter Counter Cups | Fisher Scientific | 07-000-694 | |
| Decylubiquinone | Sigma-Aldrich | D7911 | |
| DMSO | Invitrogen | D12345 | |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | 11995-065 | |
| EDTA | Sigma-Aldrich | E6758 | |
| EGTA | Sigma-Aldrich | E3889 | |
| Eppendorf Centrifuge 5425R | Eppendorf | 2231000908 | |
| Eppendorf Centrifuge 5910 Ri | Eppendorf | 5943000343 | |
| Galactose | Sigma-Aldrich | G5388 | |
| Glucose | Sigma-Aldrich | G7021 | |
| Glucose-free DMEM | Gibco | 11966025 | |
| Glutamine-free DMEM | Thermo Fisher | 11960044 | |
| Heat-Inactivated Fetal Bovine Serum | Sigma-Aldrich | F4135 | |
| Hepes | Sigma-Aldrich | H3375 | |
| HPLC-grade 35% Ammonium hydroxide | Thermo Scientific | 460801000 | |
| HPLC-grade Acetonitrile | Sigma-Aldrich | 900667 | |
| HPLC-grade Chloroform | Sigma-Aldrich | 366927 | |
| HPLC-grade formic acid | Thermo Scientific | 28905 | |
| HPLC-grade Isopropanol | Sigma-Aldrich | 563935 | |
| HPLC-grade MeOH | Sigma-Aldrich | 900688 | |
| HPLC-grade Water | Sigma-Aldrich | 270733 | |
| Human Osteosarcome Cell Line 143B | ATCC | CRL-8303 | |
| Hydrochloric Acid | Sigma-Aldrich | 320331-500ML | |
| Isotone buffer | Beckman Coulter | 8546719 | |
| K2HPO4 | Sigma-Aldrich | P2222 | |
| Mannitol | Sigma-Aldrich | M4125 | |
| MitoSox Red | Invitrogen | M36008 | |
| N-acetyl-L-cysteine | Sigma-Aldrich | A9165 | |
| Oligomycin | Sigma-Aldrich | 75351-5MG | |
| Pencillin Streptomycin | Gibco | 15140-122 | |
| Potter-Elvehjem Tissue Grinder, Size 21 | Kimble | 885502-0021 | |
| Pyruvate | Sigma-Aldrich | P5280 | |
| Pyruvate-free DMEM media | Gibco | 11965175 | |
| Q Exactive Plus Mass Spectrometer | Thermo Scientific | 726030 | |
| ReCO2ver Incubator | Baker | ||
| Refrigerated Centrivap Benchtop Vacuum Concentrator | Labconco | 7310020 | |
| RIPA Buffer | Millipore Sigma | 20188 | |
| Rotenone | Sigma-Aldrich | R8875 | |
| SeQuant ZIC-pHILIC 5μm 150 x 2.1 mm analytical column | Sigma-Aldrich | 1.50460.0001 | |
| SeQuant ZIC-pHILIC guard kit | Millipore Sigma | 1.50438.0001 | |
| Sodium Hydroxide, Pellets | Millipore Sigma | 567530-250GM | |
| Sucrose | Sigma-Aldrich | S0389 | |
| SW, TRACEFINDER 5.1 SP3 | Thermo Scientific | OPTON-31001 | |
| Tert-butyl hydroperoxide solution | Sigma-Aldrich | 458139 | |
| Tris | Sigma-Aldrich | 93352 | |
| Trypsin-EDTA (0.25%), phenol red | Gibco | 25-200-114 | |
| Uridine | Sigma-Aldrich | U3003 | |
| VANQUISH HORIZON / FLEX HPLC | Thermo Scientific | VF-S01-A-02 | |
| Z2 Coulter Particle count and size analyzer | Beckman Coulter | BZ10131270 |
Referenzen
- Spinelli, J. B., Haigis, M. C. The multifaceted contributions of mitochondria to cellular metabolism. Nature Cell Biology. 20 (7), 745-754 (2018).
- Gorman, G. S., et al. Mitochondrial diseases. Nature Reviews Disease Primers. 2, 16080 (2016).
- Chandel, N. S., et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. Journal of Biological Chemistry. 275 (33), 25130-25138 (2000).
- Cadenas, E., Boveris, A., Ragan, C. I., Stoppani, A. O. M. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome C reductase from beef-heart mitochondria. Archives of Biochemistry and Biophysics. 180 (2), 248-257 (1977).
- Murphy, M. P. How mitochondria produce reactive oxygen species. Biochemical Journal. 417 (1), 1-13 (2009).
- Schieber, M., Chandel, N. S. ROS function in redox signaling and oxidative stress. Current Biology. 24 (10), 453-462 (2014).
- Spinelli, J. B., et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science. 374 (6572), 1227-1237 (2021).
- Kumar, R., et al. A redox cycle with complex II prioritizes sulfide quinone oxidoreductase-dependent H(2)S oxidation. Journal of Biological Chemistry. 298 (1), 101435 (2022).
- Warburg, O., Geissler, A. W., Lorenz, S. On growth of cancer cells in media in which glucose is replaced by galactose. Hoppe-Seyler’s Zeitschrift für Physiologische Chemie. 348 (12), 1686-1687 (1967).
- Marroquin, L. D., Hynes, J., Dykens, J. A., Jamieson, J. D., Will, Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicological Sciences. 97 (2), 539-547 (2007).
- Attardi, G., King, M. P. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. 246 (4929), 500-503 (1989).
- Bodnar, A. G., Cooper, M., Leonard, J. V., Schapira, A. H. Respiratory-deficient human fibroblasts exhibiting defective mitochondrial DNA replication. Biochemical Journal. 305, 817-822 (1995).
- Gregoire, M., Morais, R., Quilliam, M. A., Gravel, D. On auxotrophy for pyrimidines of respiration-deficient chick embryo cells. European Journal of Biochemistry. 142 (1), 49-55 (1984).
- Mackay, G. M., Zheng, L., vanden Broek, N. J., Gottlieb, E. Analysis of cell metabolism using LC-MS and isotope tracers. Methods in Enzymology. 561, 171-196 (2015).
- Heinrich, P., et al. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Scientific Reports. 8 (1), 17910 (2018).
- Lee, P., Chandel, N. S., Simon, M. C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nature Reviews Molecular Cell Biology. 21 (5), 268-283 (2020).
- Bisbach, C. M., et al. Succinate can shuttle reducing power from the hypoxic retina to the O(2)-rich pigment epithelium. Cell Reports. 31 (2), 107606 (2020).
- Angebault, C., et al. Idebenone increases mitochondrial complex I activity in fibroblasts from LHON patients while producing contradictory effects on respiration. BMC Research Notes. 4, 557 (2011).
- Liao, P. C., Bergamini, C., Fato, R., Pon, L. A., Pallotti, F. Isolation of mitochondria from cells and tissues. Methods in Cell Biology. 155, 3-31 (2020).
- Iwai, T., et al. Sodium accumulation during ischemia induces mitochondrial damage in perfused rat hearts. Cardiovascular Research. 55 (1), 141-149 (2002).
- Murphy, E., Eisner, D. A. Regulation of intracellular and mitochondrial sodium in health and disease. Circulation Research. 104 (3), 292-303 (2009).
- Lampl, T., Crum, J. A., Davis, T. A., Milligan, C., Del Gaizo Moore, V. Isolation and functional analysis of mitochondria from cultured cells and mouse tissue. Journal of Visualized Experiments. (97), e52076 (2015).
- Murphy, M. P., et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nature Metabolism. 4 (6), 651-662 (2022).
- Xiao, Y., Meierhofer, D. Are hydroethidine-based probes reliable for reactive oxygen species detection. Antioxidants and Redox Signaling. 31 (4), 359-367 (2019).
- DeBerardinis, R. J., et al. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 104 (49), 19345-19350 (2007).
- Keeley, T. P., Mann, G. E. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiological Reviews. 99 (1), 161-234 (2019).
- Ast, T., Mootha, V. K. Oxygen and mammalian cell culture: Are we repeating the experiment of Dr. Ox. Nature Metabolism. 1 (9), 858-860 (2019).
- Gu, C., Jun, J. C. Does hypoxia decrease the metabolic rate. Frontiers in Endocrinology. 9, 668 (2018).
- Voorde, J., et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Scientific Advances. 5 (1), 7314 (2019).
- Cantor, J. R., et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell. 169 (2), 258-272 (2017).
- MacPherson, S., et al. Clinically relevant T cell expansion media activate distinct metabolic programs uncoupled from cellular function. Molecular Therapy. Methods and Clinical Development. 24, 380-393 (2022).
- Torres-Quesada, O., Doerrier, C., Strich, S., Gnaiger, E., Stefan, E. Physiological cell culture media tune mitochondrial bioenergetics and drug sensitivity in cancer cell models. Cancers. 14 (16), 3917 (2022).
- Chan, S. W., Chen, J. Z. Measuring mtDNA damage using a supercoiling-sensitive qPCR approach. Methods in Molecular Biology. 554, 183-197 (2009).
- Doherty, E., Perl, A. Measurement of mitochondrial mass by flow cytometry during oxidative stress. Reactive Oxygen Species. 4 (10), 275-283 (2017).
- Birsoy, K., et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 162 (3), 540-551 (2015).
- Beigneux, A. P., et al. ATP-citrate lyase deficiency in the mouse. Journal of Biological Chemistry. 279 (10), 9557-9564 (2004).
- Gameiro, P. A., et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metabolism. 17 (3), 372-385 (2013).
- Fendt, S. M., et al. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nature Communications. 4, 2236 (2013).
- Lee, C. P. Biochemical studies of isolated mitochondria from normal and diseased tissues. Biochimica et Biophysica Acta. 1271 (1), 21-28 (1995).
