Following this protocol, several parameters should be investigated to ensure that the tissue is correctly stained. Firstly, the TSA staining should display a good dynamic range when using low exposure times (typically 2-100 ms) during the scanning process. A low exposure time implies that the amplification has been done correctly during the reaction with HRP. For antigens stained with the secondary antibody directly coupled with the fluorochrome, the exposure time could be much longer, which could lead to photobleaching (a decrease in the signal intensity due to a long exposure time). Secondly, it is important to verify that each staining displays a high SNR. A high background signal with a low antigen signal can be an indication that the primary antibody is not specific enough, that the endogenous peroxidases were not inactivated correctly, or that one step of the protocol was not done adequately. Thirdly, depending on the slide scanner and the filter sets used for the scan, it is possible to see overlaps between two colors (e.g., AF555, AF594, and AF647). Choosing the right filter sets on the scanner and the right primary antibody dilution are crucial to avoid possible cross-detections. Quality control consists of the detection of single stained cells for each marker on the scanned file. Finally, it is also important to add a positive and negative control for each batch of staining. For immune cells, the tonsil is a good positive control. A representative result of optimal staining is shown in Figure 1.
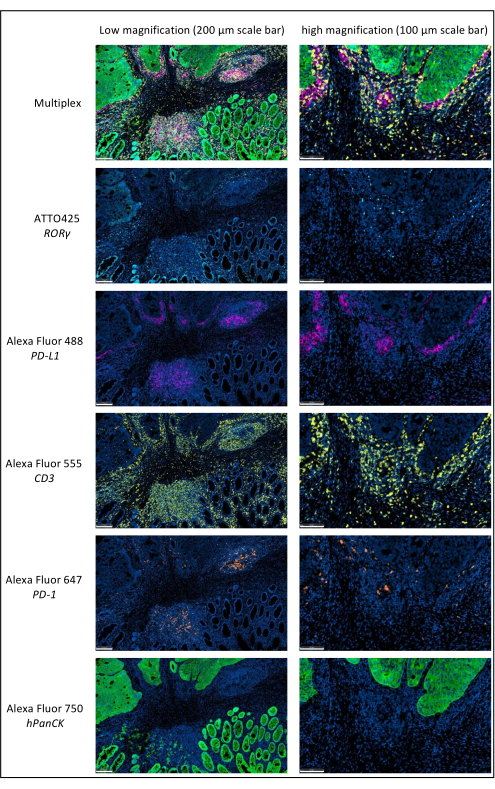
Figure 1: Locally advanced rectal cancer stained by multiplex immunofluorescence. Abbreviations: PD-1 = programmed cell death protein 1; PD-L1 = Programmed death-ligand 1; ROR-γ = RAR-related orphan receptor gamma; CD3 = cluster of differentiation 3; hPanCK = human pan-cytokeratin. Each antigen staining is scanned in grayscale, and the colors presented in the figure are pseudocolors. Scale bar low magnification: 200 µm; scale bar high magnification: 100 µm. Please click here to view a larger version of this figure.
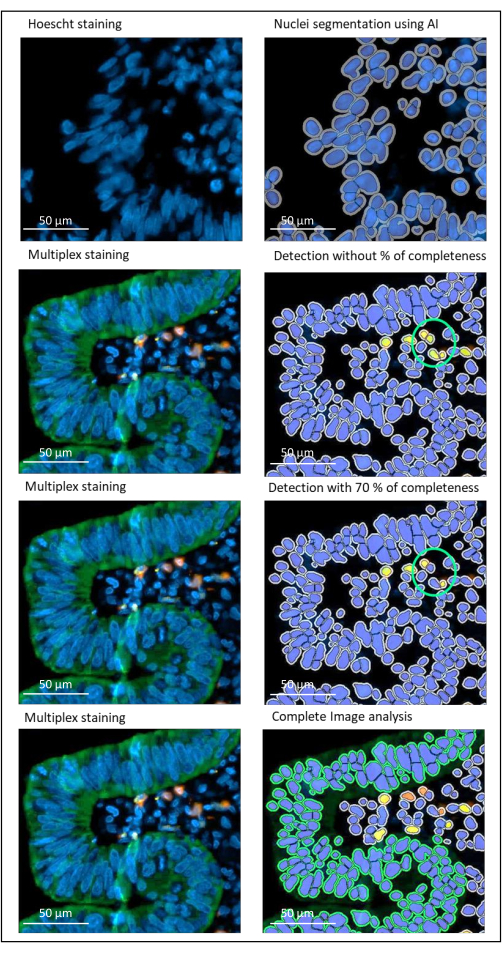
Figure 2: Nuclei and staining detection of a locally advanced rectal cancer using an image analysis software. Without the percentage of completeness parameter correctly set, the software detects two CD8+ cells (green circle) because they are close to each other, but only one cell is stained. Using 70% completeness helps to avoid this false positive detection. Green = hPanCK; Yellow = CD3; Orange = CD8. Scale bar: 100 µm Please click here to view a larger version of this figure.
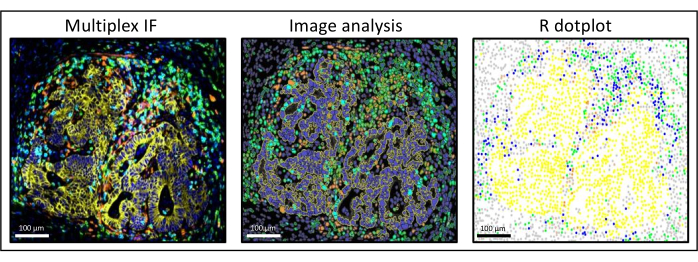
Figure 3: Image analysis and R dot-plot reconstitution of a liver colorectal cancer metastasis. On the multiplex staining (left), human pan-cytokeratin is in yellow, CD3 is in green, CD8 is in light blue, and IDO is in orange. On the dot-plot (right), human pan-cytokeratin+ cells are in yellow, CD3+CD8− cells are in green, CD3+CD8+ cells are in blue, and IDO+ cells are in orange. Please click here to view a larger version of this figure.
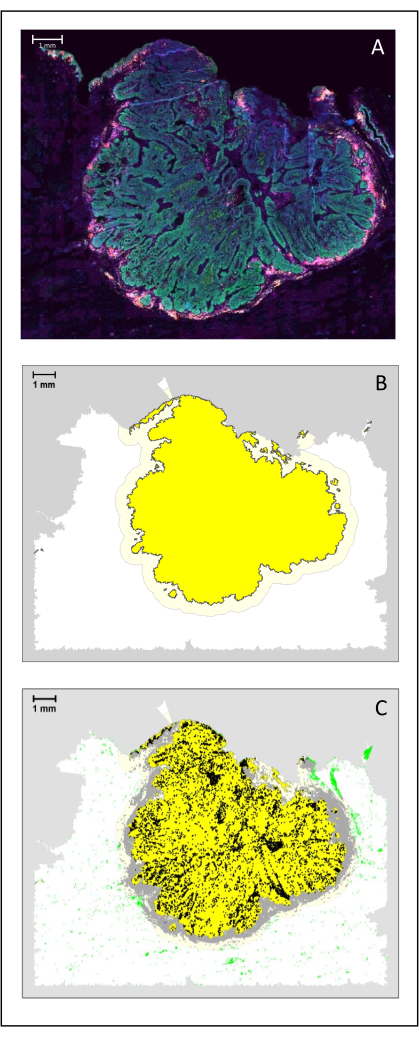
Figure 4: Analysis of a surgical section of an HNSCC. (A) A surgical section of an HNSCC. Cancer cells are visible in green. Peri-tumoral cells are visualized around the tumor islets (CD3 in yellow and CD8 in purple). (B) The center of the tumor (in yellow with a black border) is computed bioinformatically by the k-nearest-neighbor algorithm based on the distance between the tumor islands from a single area. Around this area, an invasive margin (light yellow with a grey border) is computed on an arbitrary 500 µm basis. (C) Invasive T cells are highlighted with black dots in the center of the tumor and grey dots in the invasive margin. Other T cells are highlighted in light green dots. Scale bar: 1 mm. Please click here to view a larger version of this figure.
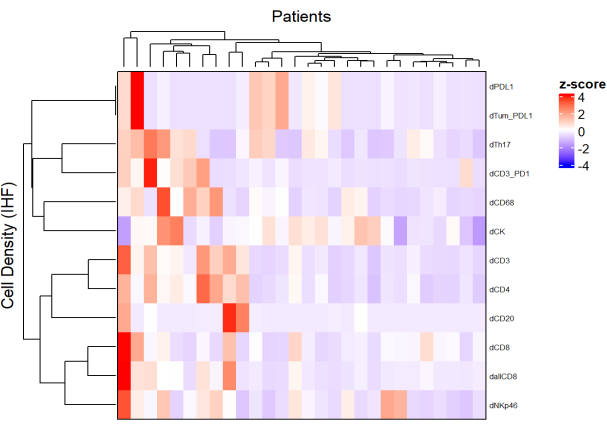
Figure 5: Heatmap of the density of different cell types of locally advanced rectal cancer biopsies. The heatmap was drawn using unsupervised clustering of the densities of different cell types from different multiplex panels with the ComplexHeatmap package. Scaling and centering were used for normalization. Please click here to view a larger version of this figure.
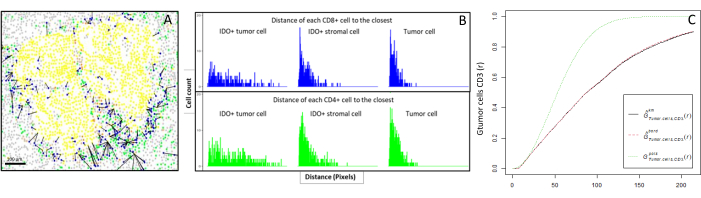
Figure 6: Distances of the CD4+ and CD8+ cells to each IDO+ or tumoral cell. Human pan-cytokeratin+ cells are in yellow, CD3+CD8− cells are in green, CD3+CD8+ cells are in blue, and IDO+ cells are in orange. (A) The closest distance between tumor cells and each CD8+ T cell. (B) Barplots of the distances between IDO+ cells and each CD8+ T cell (blue) or CD4+ T cell (green). (C) Example of a sample analyzed by the G-cross function. The y-axis shows the probability of a tumor cell encountering a CD3+ lymphocyte in a radius ranging from 0-200 µm around the tumor cell. Three curves are shown; the theoretical curve is in dotted green (Poisson distribution), the corrected empirical curve with km correction is in black, and the corrected empirical curve with border correction is in dotted red. Please click here to view a larger version of this figure.
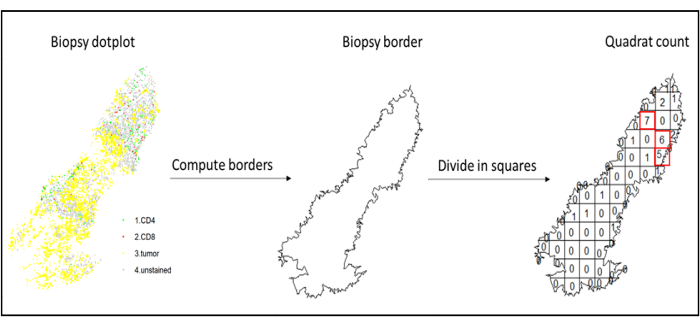
Figure 7: Illustration of a Quadratcount. Border computation and quadratcount were performed using the spatstats package. The most infiltrated squares (hotspots) can be used for downstream statistics. CD4 is in green, CD8 is in red, and tumor cells are in yellow. Please click here to view a larger version of this figure.
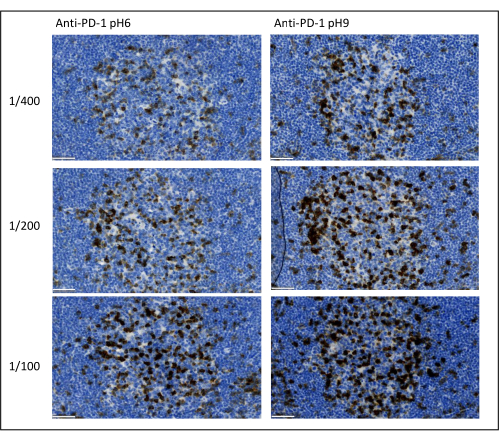
Figure 8: Antibody dilution and antigen retrieval optimization. Chromogenic detection of PD-1 using three different dilutions and two different antigen retrieval solutions of the primary antibody (Citrate pH 6 and EDTA pH 9). Scale bar: 50 µm. Please click here to view a larger version of this figure.
| Primary antibody | Dilution | Antigen retrieval | Secondary antibody | Fluorochrome | Position |
| PD-1 | 1/100 | EDTA (pH 9) | Anti-rabbit | AF647 | 1 |
| PD-L1 | 1/1000 | EDTA (pH 9) | Anti-rabbit | AF488 | 2 |
| ROR-γ | 1/200 | EDTA (pH 9) | Anti-mouse | ATT0-425 | 3 |
| CD3 | 1/100 | Citrate (pH 6) | Anti-rabbit | AF555 | 4 |
| hPanCK | 1/50 | Citrate (pH 6) | Anti-mouse coupled with AF750 | 5 | |
Table 1: Example of an optimized multiplex panel. Abbreviations: PD-1 = programmed cell death protein 1; PD-L1 = Programmed death-ligand 1; ROR-γ = RAR-related orphan receptor gamma; CD3 = cluster of differentiation 3; hPanCK = human pan-cytokeratin; AF = AlexaFluor; EDTA = ethylenediaminetetraacetic acid. CD3 is used to detect T lymphocytes; PD-1 is used to detect exhausted lymphocytes; ROR- γ is used to detect Th-17; and hPanCK is used to detect tumor cells. The position column indicates the order in which the sequential multiplex has to be performed.
