liCHi-C offers the possibility of generating high-quality and resolution genome-wide promoter interactome libraries with as little as 50,000 cells53. This is accomplished by – besides the drastic reduction of reaction volumes and the use of DNA low-binding plasticware throughout the protocol – removing unnecessary steps from the original protocol, in which significant material losses occur. These include the phenol purification after decrosslinking, the biotin removal, and subsequent phenol-chloroform purification and ethanol precipitation. Besides that, reorganizing the steps of the Hi-C library preparation (biotin pulldown, A-tailing, adapter ligation, PCR amplification, and double-sided paramagnetic bead selection-also as the PCR product purification) allows us to remove yet another unnecessary DNA purification step. An overview of the experimental workflow can be found in Figure 1A.
To assess library quality, several controls throughout the protocol are performed, the first of which is the calculation of genome digestion efficiency; values over 80% are considered acceptable (Table 3). Checking for the digestion efficiency of the cell type in a separate experiment is suggested in order to not lose a significant amount of material from a single liCHi-C experiment. Second, before sonication and end-repair, it is recommended to check for the sensitivity of interaction detection by amplifying cell-type invariant chromatin interactions by conventional PCR (Figure 1B). If the specific product is detected, a third control should be performed, focusing on the efficiency of biotinylation and ligation by differentially digesting one of the previously obtained PCR products with HindIII and NheI (Figure 1C,D). When filling a digested HindIII restriction site and blunt-end ligating it with another one, a new NheI restriction site is generated instead of regenerating the original HindIII one. Therefore, the digestion of the PCR amplicon should only be observed when NheI is present. Finally, just before and after the entire capture, the concentration and size distribution should be checked using automated electrophoresis. Pre-capture library amplification must aim at obtaining 500-1,000 ng of Hi-C material, the exact amount needed to perform the RNA probe capture, since excessive amplification of both the pre- and post-captured library leads to a high percentage of PCR duplicates and the consequent loss of sequencing reads during analysis. Libraries can be reamplified again under the same conditions if not enough material is obtained during the first conservative PCR amplification. The amount of post-captured library material can vary, but as a rule of thumb, it should be approximately tenfold to 20-fold less than before the capture. The size distribution of the library should fall around 450-550 bp (Figure 2A,B), invariable between pre- and post-captured libraries. Collectively, correct results of these controls ensure the generation of excellent liCHi-C libraries.
Finished liCHi-C libraries are then (at least) 100 bp paired-end sequenced and analyzed. Raw sequencing data53 is processed using the HiCUP pipeline56 for mapping and filtering out artifacts. The ideal HiCUP report shows a fivefold to tenfold increase in the distribution of cis (inside the same chromosome) compared to trans (between different chromosomes) paired-end reads, as described previously according to in-nucleus ligation Hi-C57 (Figure 3B). The obtention of more than 100 million unique, valid reads after the removal of PCR duplicates is enough to proceed to the following step in the analysis (Figure 3B), which is to assess the capture efficiency. Paired-end reads in which none of their ends map into a captured restriction fragment by the RNA probe enrichment system, are discarded, keeping only the ones representing the promoter interactome of the cell (i.e., those reads in which at least one of their ends maps into restriction fragments containing one or more gene promoters [Figure 3C], ideally more than 60%).
Finally, significant interactions are called with the CHiCAGO pipeline, as described in58,59. Two or more biological replicates are needed for the final set of significant promoter interactions. The data quality can also be validated using principal component analysis (PCA), since biological replicates must cluster together and cell types must be separated. For instance, by analyzing liCHi-C datasets from four different primary cell types from the human hematopoietic tree (common myeloid progenitors, monocytes, megakaryocytes, and erythroblasts), we can observe in a PCA that liCHi-C libraries cluster in a developmental trajectory-reflecting fashion (Figure 3D). A closer examination of the significant interactions detected for the four cell types reveals that promoter interactomes are cell-type specific and dynamic during cell development. For example, the AHSP gene, a key chaperone synthesized in erythroid precursors which oversees the correct folding of hemoglobin60,61,62, shows a gain of interactions with potentially active regulatory elements (i.e., H3K27ac and H3K4me1 enriched regions) in erythroblasts, but not in other cell types (Figure 4). This demonstrates that the liCHi-C method can uncover potential regulatory interactions in rare cell types.
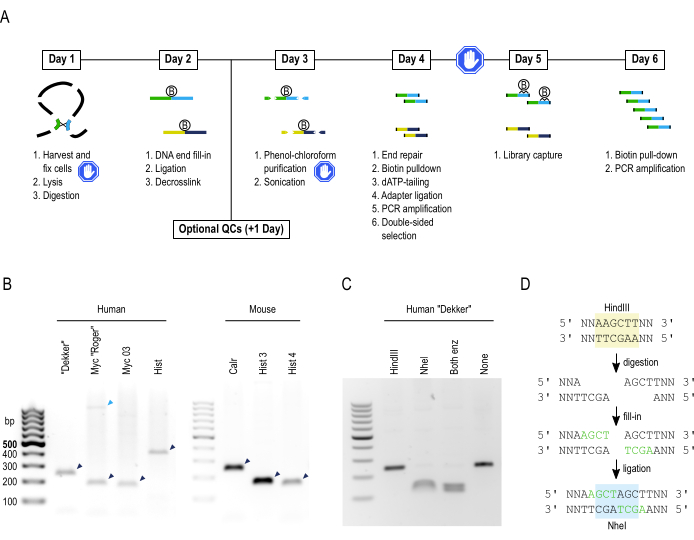
Figure 1: Protocol overview and quality controls of the sample before sonication. (A) Schematic overview of the liCHi-C protocol divided by days. B and blue hands represent, respectively, biotin molecules and steps in which one can safely stop the protocol for a large period of time. (B) Representative results of the 3C interaction controls. Both interaction sets for human (left) and mouse (right) are shown. The bands to expect are marked in dark blue, while an unspecific band is marked in light blue. (C) Representative fill-in and ligation controls using the "Dekker" human interaction primer pair. The band is cut only in lanes 2 and 3, where NheI is added. (D) Schematic representation of the generation of a new NheI restriction site during fill-in and ligation of a HindIII restriction site. Please click here to view a larger version of this figure.
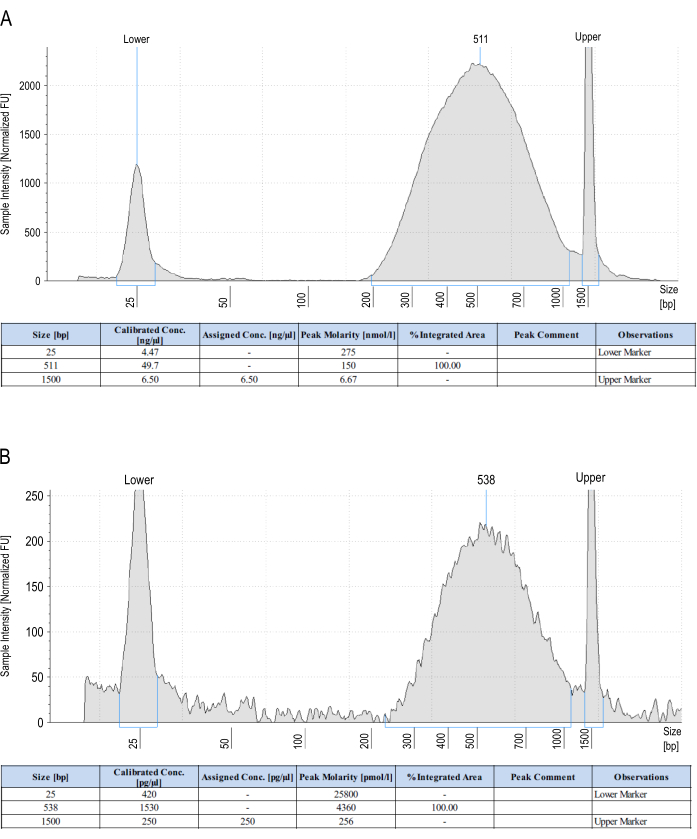
Figure 2: Representative automated electrophoresis profiles from libraries pre- and post-capture. (A) Automated electrophoresis profile from a library just before capture. The total amount of DNA obtained is 994 ng (49.7 ng/µL in 20 µL). (B) High-sensitivity automated electrophoresis profile from a finished liCHi-C library. The sample is loaded half-diluted to preserve as much material as possible. The total amount of DNA obtained is 61.2 ng (1.53 ng/µL in 20 µL x2 to account for the dilution). Please click here to view a larger version of this figure.
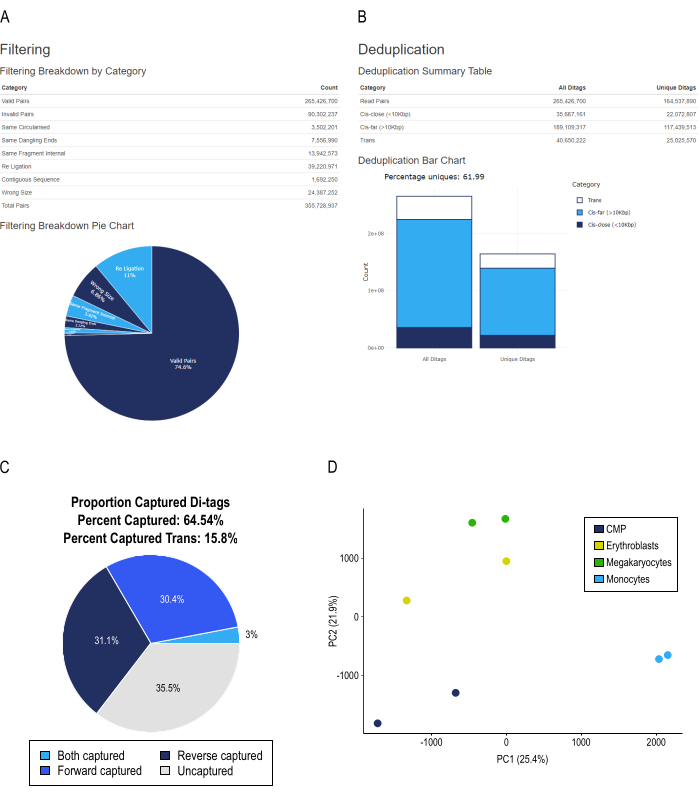
Figure 3: Representative HiCUP pipeline output and sample replicate clustering by PCA. (A) Classification of the validity of read pairs by percentages and total counts. Invalid read pairs are subclassified by the experimental artifact type. (B) Deduplication percentages and classification of the interaction types. Cis interactions are further subclassified as cis-close (less than 10 kb) and cis-far (more than 10 kb). (C) Percentage of capture efficiency. Captured read-pairs are further subclassified whether one end, the other, or both contain one or more promoters. (D) Principal component analysis of CHiCAGO significant interaction scores from both replicates of liCHi-C libraries from common myeloid progenitors (CMP), erythroblasts, megakaryocytes, and monocytes. Please click here to view a larger version of this figure.
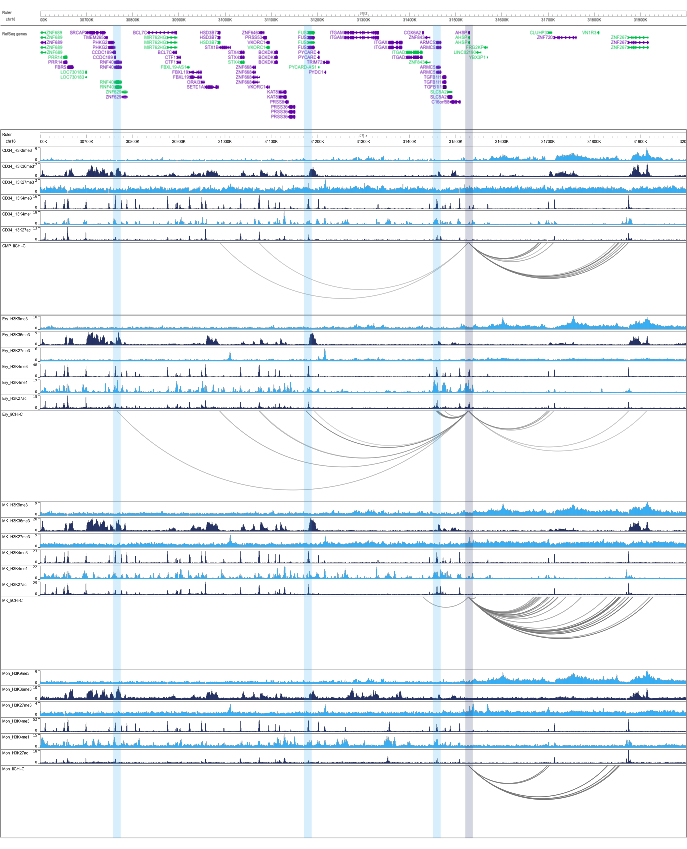
Figure 4: AHSP interaction landscape in human primary hematopoietic cells. Representative example of the AHSP promoter-centered interaction landscape in common myeloid progenitors (CMP), erythroblasts (Ery), megakaryocytes (MK), and monocytes (Mon) as seen in the WashU Epigenome Browser63. Arcs represent significant interactions. The dark blue shade shows the AHSP gene promoter, while the light blue shades overlap potential active regulatory elements that interact specifically with the AHSP promoter in erythroblasts. Please click here to view a larger version of this figure.
Table 1: Buffer composition and preparation. Please click here to download this Table.
Table 2: PCR primers and adapter sequences. Please click here to download this Table.
Table 3: Example of the calculation for the digestion efficiency. Please click here to download this Table.
