Podocytes are specialized, post-mitotic renal epithelial cells that form the glomerular filtration barrier of the kidney along with the glomerular basement membrane (GBM), glomerular endothelial cells, and glycocalyx. Phenotypically, podocytes consist of a cell body and primary, microtubule-driven membrane extensions, as well as secondary extensions called foot processes1,2. The glomerular filtration barrier that filters urine from the blood is built of fenestrated endothelium, GBM, and a specialized type of intercellular junction that connects neighboring podocyte foot processes, called the slit diaphragm of podoctyes3. Under healthy conditions, proteins larger than albumin are retained from the filtration barrier due to their size and charge4.
Mutations in cytoskeletal- or podocyte-specific genes, as well as circulating factors affecting podocyte signaling pathways, are known to induce podocyte effacement, detachment, or apoptosis, resulting in proteinuria and glomerular sclerosis. In particular, cytoskeletal rearrangement, changes in podocyte polarity or damage of foot processes with an associated loss of slit junctions are pivotal5. Due to their terminally differentiated status, podocytes can hardly be replaced after detachment of the GBM. However, if podocytes are attached to the GBM, they can still recover from effacement and reform interdigitating foot processes6,7,8. Further understanding of the events that lead to podocyte damage in various glomerular disorders may provide novel therapeutic targets that will aid in developing treatments for these diseases. Podocyte damage is a hallmark of different glomerular diseases, including focal segmental glomerulosclerosis (FSGS), diabetic nephropathy, minimal change disease, and membranous glomerulonephropathy, requiring reliable podocyte ex vivo models to study the pathological mechanisms and potential treatment approaches for these diseases9,10. Podocytes can be studied ex vivo by classical primary cell culture based on the isolation of glomeruli by differential sieving11. However, due to the terminally differentiated state with limited proliferation capacity, most researchers use mouse or human ciPodocyte cell lines that express temperature-sensitive variants of the SV40 large T antigen. Alternatively, ciPodocytes are isolated from transgenic mice harboring the SV40 Tag immortalizing gene1,12.
CiPodocytes proliferate at 33 °C, but enter growth arrest and start differentiating at 37 °C13,14. It has to be kept in mind that experimental data obtained with these cells should be interpreted with certain caution, as the cells are generated using an unnatural gene insertion15. As these cells harbor an immortalizing gene, the cellular physiology is altered because of ongoing proliferation12. Podocyte cell lines generated by this approach have recently been questioned, as mouse, human, and rat ciPodocytes express less than 5% of synaptopodin and nephrin at the protein level, as well as NPHS1 and NPHS2 at the mRNA level compared to glomerular expression16. Moreover, most podocyte cell lines do not express nephrin17,18. Chittiprol et al. also described a significant difference in cell motility and responses to puromycin and doxorubicin in ciPodocytes16. Podocytes can be found in urine after detachment from the GBM in different glomerular diseases19,20,21,22. Viable urinary podocytes can be cultivated ex vivo for up to 2-3 weeks, but most cells undergo apoptosis23,24. Interestingly, podocytes are not only found in the urine of patients with glomerular disease but also in the urine of healthy subjects, most likely when they are senescent-again with a limited potential of replication in culture24. Furthermore, the urinary-derived podocyte number is limited, and the cells dedifferentiate in culture, show less foot processes, change morphology, and most importantly have limited proliferation capacity. The expression of podocyte-specific genes is absent, disappears within a few weeks, or varies among these cell clones. Some cells positive for the podocyte-specific marker co-expressed the marker of tubular epithelial cells or myofibroblasts and mesangial cells, which suggests dedifferentiation and/or transdifferentiation of the cultured urinary podocytes24,25.
Recently, the generation of ciPodocyte cell lines derived from the urine of patients and healthy volunteers by transduction with a thermo-sensitive SV40 large T antigen and hTERT has been described26. mRNA expression for synaptopodin, nestin, and CD2-associated protein was detected, but podocin mRNA was absent in all clones. In addition to the problems with urinary podocytes, these cells also contain the inserted immortalizing gene, resulting in the disadvantages discussed above.
In contrast, human induced pluripotent stem cells (hiPSCs) have a huge capacity to self-renew and differentiate into multiple cell types under appropriate conditions. It has been shown before that hiPSCs can serve as an almost unlimited source of podocytes27,28.
Here, a two-step protocol to generate patient-specific podocytes from dermal fibroblasts of skin punch biopsies with subsequent episomal reprogramming into hiPSCs and a final differentiation into hiPSC-derived podocytes is described (Figure 1).
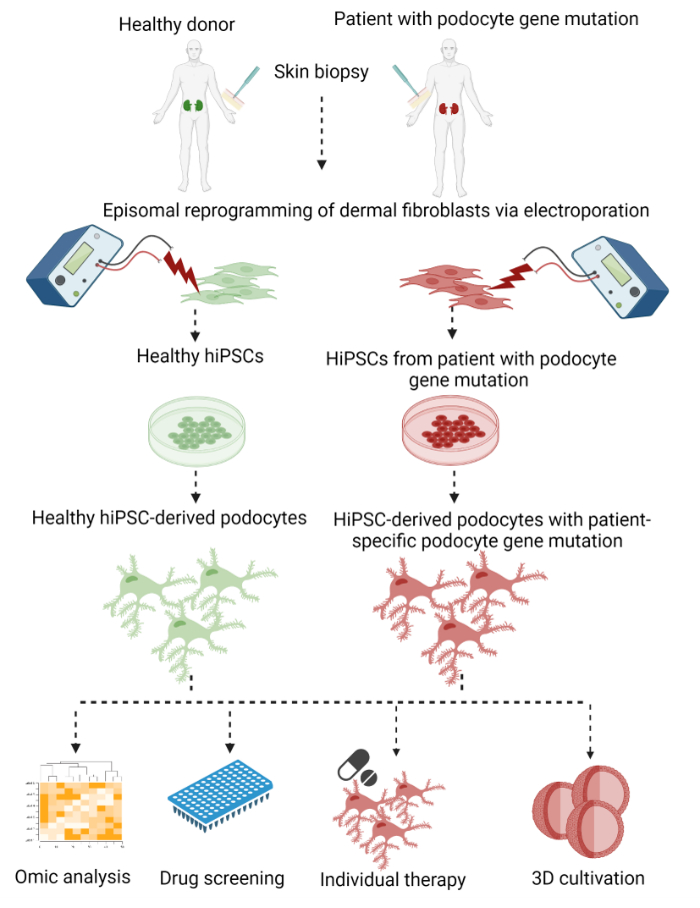
Figure 1: Protocol to generate patient-specific hiPSC-derived podocytes. Graphical overview of the protocol to generate patient-specific podocytes from dermal fibroblasts of a skin biopsy by reprogramming into hiPSCs and differentiation into podocytes. Please click here to view a larger version of this figure.
As a first step, somatic dermal fibroblasts were outgrown from a skin punch biopsy and reprogrammed into hiPSCs using an integration-free method by electroporation with plasmids expressing the transcription factors OCT3/4, KLF4, SOX2, and c-MYC29,30,31. Arising hiPSC colonies were subsequently selected and expanded. Differentiation began with the induction of the mesodermal lineage by activation of the WNT signaling pathway, followed by the generation of nephron progenitor cells that were still able to proliferate. Finally, the cells were differentiated into podocytes. In this procedure, we modified and combined previously published protocols for episomal reprogramming for the generation of hiPSCs by Bang et al.32 and Okita et al.33, as well as a protocol for the differentiation of hiPSCs into podocytes by Musah et al.28,34,35.
Indeed, podocytes generated by our protocol had a phenotype closer to podocytes in vivo, regarding the development of a distinct network of primary and secondary foot processes and the expression of podocyte-specific markers, like synaptopodin, podocin, and nephrin. With the use of hiPSC-derived podocytes, the patient's genetic background is maintained during reprogramming and differentiation. This enables patient-specific podocyte disease modelling and the discovery of potential therapeutic substances ex vivo in an almost unlimited cell number. Moreover, this protocol is minimally invasive, cost-efficient, ethically acceptable and may facilitate new avenues for drug development.
With this step-by-step protocol that combines episomal reprogramming and differentiation, it is possible to generate podocytes carrying a patient's mutation in a podocyte-relevant gene. This enables the analysis of disease-specific podocyte alterations ex vivo. The patient's genetic background is preserved during the protocol during all the different cell stages. In addition, the limitation of insufficient cell number of terminally differentiated non-proliferating podocytes can be overcome by using hiPSC-derived podocytes. Although it takes several months until hiPSC-podocytes are generated from outgrown dermal fibroblasts via reprogramming into hiPSCs and subsequent differentiation into podocytes, it is possible to freeze cells at three different steps of the protocol (Figure 2). Freezing is possible for fibroblasts, hiPSCs, and nephron progenitor cells after differentiation in nephron progenitor differentiation medium for 7 days. Therefore, the generation of working cell banks and large-scale experiments is possible.
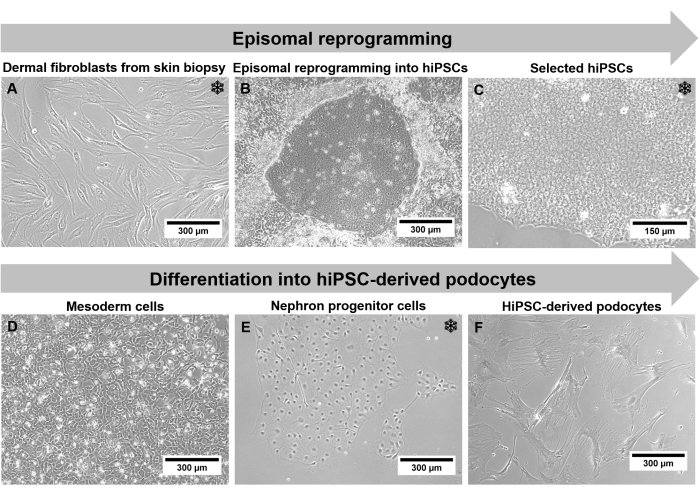
Figure 2: Individual steps of the protocol through a phase contrast microscope. (A) Outgrown dermal fibroblasts. (B) hiPSC colony formation after reprogramming. (C) Selected hiPSC culture. (D) Mesoderm cells after 2 days of differentiation. (E) Nephron progenitor cells after 14 days of differentiation in nephron progenitor differentiation medium. (F) Terminal differentiated hiPSC-derived podocytes. Scale bars represent 300 µm (A,B,D-F) and 150 µm (C). The snowflake highlights possible freezing steps. Please click here to view a larger version of this figure.
As the generated hiPSC-derived podocytes traverse a long protocol with drastic changes regarding cell type and morphology, cell characterization is obligatory. Cell morphology, like size and cell shape, as well as growth behavior, can be monitored using a phase contrast microscope. Fibroblasts present a long spindle-like phenotype with a size of 150 µm to 300 µm (Figure 2A). After reprogramming, colonies with 50 µm small hiPSCs occur. These colonies display distinct borders and hiPSCs distinguished with a high nuclei-to-body ratio and increased proliferation rate (Figure 2C). Since podocytes are terminally differentiated cells, the proliferation rate decreases with progressive differentiation, and the cell morphology changes to 300 µm large star-shaped podocytes with prominent foot processes (Figure 2F).
Confluency is a critical point of hiPSC culture, and spontaneous differentiation can appear when colonies grow too dense (Figure 3). These colonies must be removed before passaging by scraping the affected parts of the culture dish.
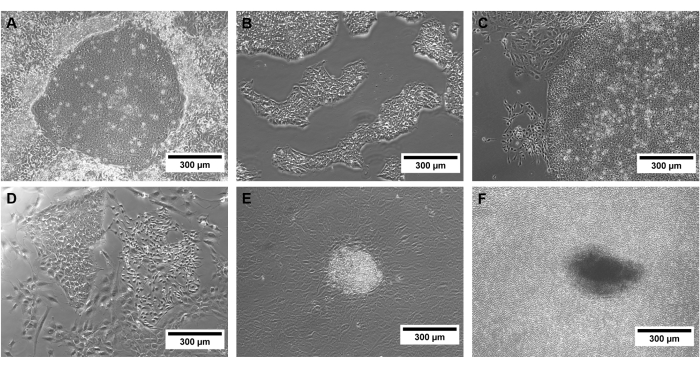
Figure 3: Examples of hiPSCs of different quality. Phase contrast images of successfully (A) reprogrammed hiPSC colonies and (B) selected hiPSCs, as well as (C) spontaneous differentiation, (D,E) non-hiPSC colonies, and (F) a too-dense hiPSC culture. Scale bars represent 300 µm. Please click here to view a larger version of this figure.
The different cell types of this protocol must be validated regarding the expression of a specific marker. After reprogramming, hiPSCs regain pluripotency capacity and express proliferation and pluripotency markers like SSEA4, OCT3/4, and Ki67 (Figure 4)38,39,40. It is known that reprogramming, as well as the lengthy culture of hiPSCs and differentiation, can lead to an abnormal karyotype, or rather induce mutations41. Therefore, the genetic background of the cultured cells should be monitored over time and at different passages by g-banding and whole exome sequencing.
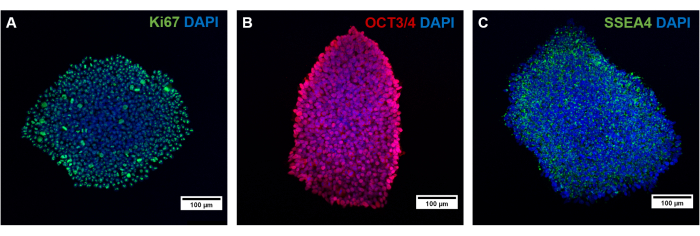
Figure 4: Characterization of generated hiPSCs by immunofluorescence staining. Characterization of generated hiPSCs by staining for (A) the proliferative marker Ki67, as well as the pluripotency markers (B) OCT3/4 and (C) SSEA4. Scale bars represent 100 µm. Please click here to view a larger version of this figure.
Morphologically, hiPSC-derived podocytes appear star-shaped and express a distinct network of long primary and secondary foot processes compared to ciPodocytes (Figure 5). HiPSC-derived podocytes express podocyte-specific marker proteins like synaptopodin, nephrin, and podocin (Figure 6A–C)42.
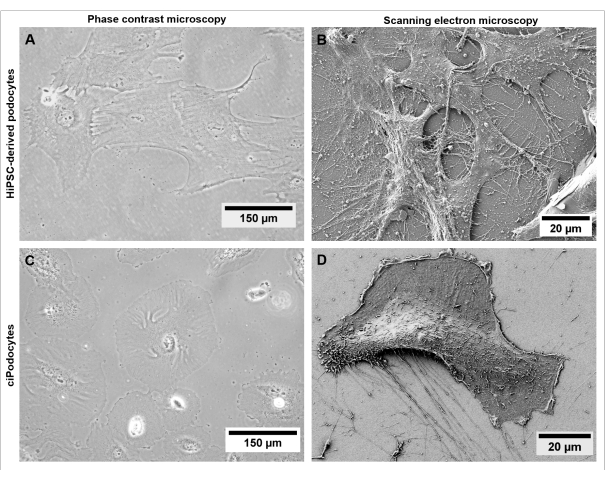
Figure 5: Morphology of hiPSC-derived podocytes compared to ciPodocytes. Comparison of (A,B) hiPSC-derived podocytes from a healthy donor to (C,D) ciPodocytes regarding cell morphology and filopodia by using a phase contrast microscope (A,C) and scanning electron microscope (B,D). Scale bars represent 150 µm (A,C) and 20 µm (B,D). Please click here to view a larger version of this figure.
Therefore, the comparison of hiPSC-derived podocytes not only from healthy donors, but also from patients with mutations in podocyte-specific genes, allows for individualized characterization of the patient's mutation ex vivo.
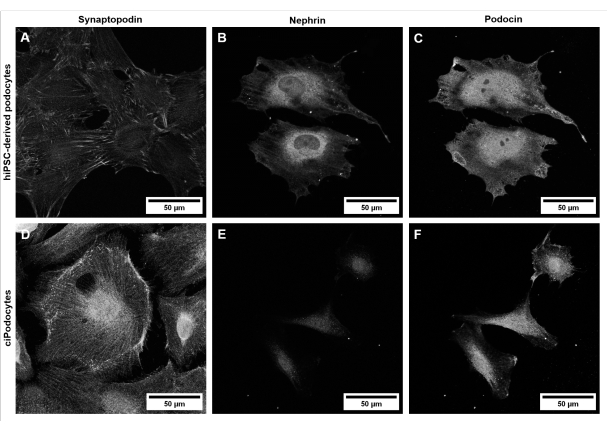
Figure 6: Characterization of a podocyte-specific marker in hiPSC-derived podocytes and ciPodocytes. Comparison of (A–C) hiPSC-derived podocytes from a healthy donor to (D–F) ciPodocytes regarding podocyte-specific marker proteins synaptopodin (A,D), nephrin (B,E), and podocin (C,F). Scale bars represent 50 µm. Please click here to view a larger version of this figure.
Table 1. Composition of all cell culture media and solutions used in the study. Please click here to download this Table.
