Vascular niche cultures were established by sequentially seeding hBM-MSCs and GFP-HUVECs onto pre-cast PEG-based hydrogels with a stiffness gradient within a 96-well imaging plate (Figure 1). The cultures were monitored longitudinally via live epifluorescence microscopy and further characterized at selected time points. The extracellular compartment was assessed via direct color stainings and antibody-based stainings. ALP activity was quantified after retrieving and lysing the cells from the generated niches. Furthermore, we demonstrate the suitability of this platform for anti-angiogenic drug sensitivity assays and as a basis for cancer co-culture models.
Co-cultures of hBM-MSCs and GFP-expressing HUVECs were established by seeding 3 x 104 cells/well in the absence of growth factors or in the presence of either FGF-2 or BMP-2 at 50 ng/mL, as described in the protocol. From an early time point, differences between the cultures could be observed both from the bright-field and fluorescence images showing only the GFP-HUVECs (Figure 2A). By observing the same areas longitudinally, differences in the development of the cultures could be noted, such as a faster development in the presence of FGF-2. Generally, the cultures appeared less developed in the absence of any growth factor, with fewer cells of either type spreading and the presence of acellular areas. In contrast, the bright-field images showed the most dense cultures in the presence of BMP-2. However, vascular-like networks formed in both growth factor-containing conditions, and the most extensive and interconnected networks were formed with FGF-2. These observed differences could also be quantified using Angiogenesis Analyzer for ImageJ. Indeed, the total network length was highest in the presence of FGF-2 and lowest in the absence of growth factors (Figure 2B). The number of junctions indicating branching points in the networks followed the same trend as the total length (Figure 2C). Conversely, both growth factor-containing conditions featured significantly fewer isolated segments, indicating higher interconnectivity, than the condition without any growth factor (Figure 2D). Additionally, 3D confocal imaging revealed stronger endothelial cell penetration in terms of depth in the FGF-2-stimulated condition (Figure 2E).
hBM-MSC/GFP-HUVEC co-cultures were maintained for 7 days in the presence of FGF-2 or BMP-2 before being fixed and stained for ECM components. Immunocytochemical stainings followed by confocal laser scanning microscopy showed striking differences in culture morphology depending on the type of growth factor supplementation (Figure 3A). With FGF-2, the culture was organized into condensed microvascular-like structures, which were dense in both endothelial and mesenchymal cells, whereas in the presence of BMP-2, the hBM-MSCs spanned a much larger area, as evident from the more extensive F-actin-positive and GFP-negative areas. The ECM proteins fibronectin and collagen I were localized in a similar manner, while laminin and collagen IV were more concentrated around the endothelial structures. However, this increased concentration around the endothelial structures was much more pronounced in the presence of FGF-2 than in the presence of BMP-2. In addition to the antibody-based stainings, direct color stainings were performed to assess the overall fibrotic state of the ECM (Picrosirius Red staining; Figure 3B), as well as the deposition of Ca on the ECM (Alizarin Red staining; Figure 3C) of the formed niches. The Picrosirius Red staining was stronger and more extensive in the niches cultured with BMP-2, and the Alizarin Red staining followed the same trend.
Next, the cultures were characterized in terms of their osteogenic potential by assessing ALP activity as an early osteogenic marker. On day 4 and day 7 of co-culture, cells were retrieved from the niches by digesting the hydrogels with Trypsin. To quantify the ALP activity, the retrieved cells were lysed, and a pNPP assay was performed. The values obtained were normalized against the total DNA for each sample to account for potential differences in the cell numbers across conditions. Indeed, small differences between the DNA content of the conditions could be observed, and the fewest cells were retrieved from the condition without any growth factor (not shown). The normalized ALP activity, however, varied greatly across conditions, with a trend of increasing activity with higher concentrations of BMP-2 and a plateau at 100 ng/mL (Figure 4A,B). The lowest activity levels were identified for the condition containing 50 ng/mL FGF-2. While similar trends could be observed for both of the time points assessed, all the values significantly increased over time in culture from day 4 to day 7. In addition to the quantitative assay, the ALP activity could be qualitatively visualized using direct color staining based on the BCIP/NBT substrate conversion. More extensive and more intense purple staining was observed in the presence of BMP-2 as compared to FGF-2 (Figure 4C).
To demonstrate two potential applications of the characterized osteogenic niches, a proof-of-concept drug sensitivity study and cancer co-cultures were performed. For the drug sensitivity assay, bevacizumab or a control solution consisting of the diluent of the bevacizumab formulation was added to the culture medium containing fresh BMP-2. On day 4, when established networks were formed in the presence of 50 ng/mL BMP-2, either the control solution or bevacizumab-containing medium was added during the regular medium change, and the cultures were monitored using fluorescence imaging. The addition of 10 µg/mL bevacizumab led to the retraction or ablation of the previously formed network, while the control condition still featured extensive networks 2 days after the medium change (Figure 5A,B). These changes could also be quantified by tracking the total length of the networks using Angiogenesis Analyzer for ImageJ on fluorescence images acquired daily (Figure 5C). Alternatively, bevacizumab or any other compound could also be added from the beginning of the co-culture to assess their influence on the formation of the networks. In the case of bevacizumab, this completely inhibited the formation of endothelial networks (not shown).
For the second application, MDA-MB-231 breast cancer or U2OS osteosarcoma cells were added to day 4 co-cultures at a density of 1.5 x 103 cells/well in fresh culture medium containing 50 ng/mL BMP-2. To distinguish them from the GFP-labelled HUVECs and the non-labeled hBM-MSCs, the cancer cells were incubated with CellTrace FarRed just before the seeding onto the osteogenic niches. The cultures were monitored by fluorescence microscopy; at the beginning, most of the cancer cells localized near the surface of the substrate, but after 2 days, they could be found in closer proximity to the layers containing the vascular co-cultures. Thus, day 2 was selected as the starting point for time-lapse microscopy to show the dynamics of the interactions between the cancer cells and the cells within the vascular niche. Interestingly, the MDA-MB-231 cells could be seen both approaching and moving away from endothelial structures and, thus, were possibly probing or remodeling their environment (Figure 5D). Using CellTrace FarRed as a label for the cancer cells, GFP as a label for HUVECs, and additional staining for F-actin, all the cell types could be distinguished using confocal laser scanning microscopy (Figure 5E).
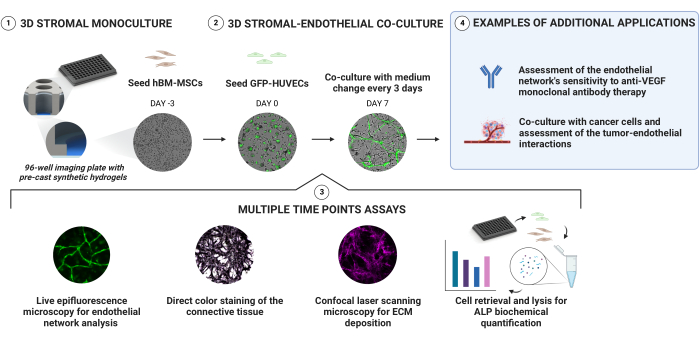
Figure 1: A simple approach for the reliable generation of vascularized, osteogenic niches. Pre-cast synthetic hydrogels with an in-depth stiffness gradient allow for the generation of 3D cultures via sequential cell seeding without the need for direct encapsulation. hBM-MSCs are pre-cultured for 3 days before GFP-expressing HUVECs are added. The cultures are monitored by acquiring bright-field and GFP signals longitudinally. At select time points, the niches are further evaluated for their ECM deposition and osteogenic state. Alkaline phosphatase activity is assessed via direct color staining and by retrieving cells from the niches and performing a pNPP assay on the cell lysates. Please click here to view a larger version of this figure.
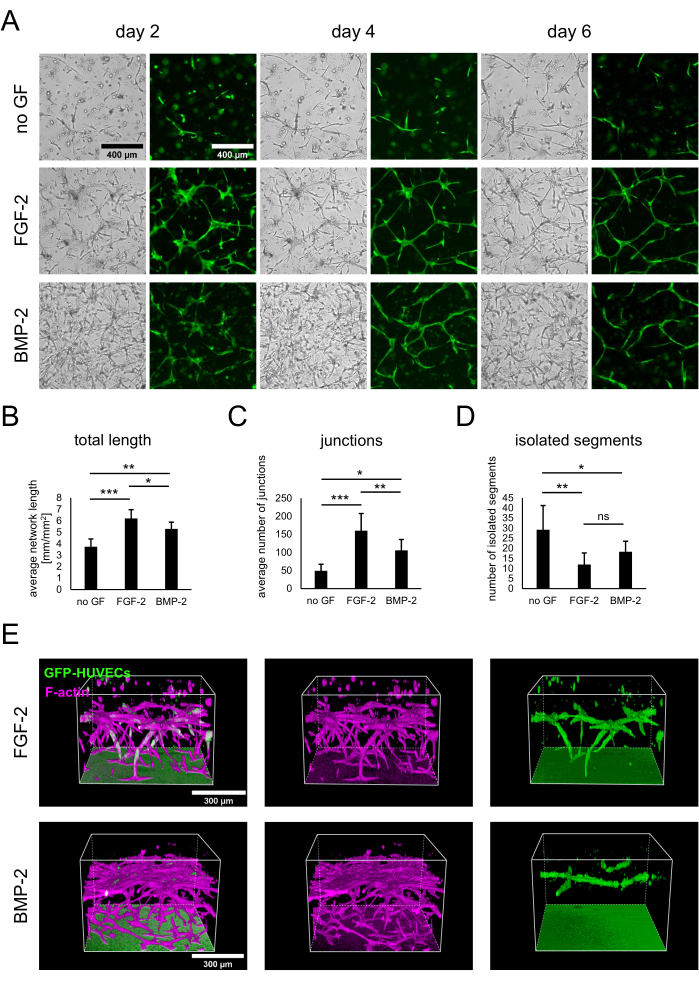
Figure 2: Stimulation of vascular-like networks with FGF-2 or BMP-2. (A) Bright-field and fluorescence (GFP) images were acquired on day 2, day 4, and day 6 of the co-culture of cells grown in the absence of growth factors or in the presence of FGF-2 or BMP-2, both at 50 ng/mL. Scale bar: 400 µm. (B–D) Quantified parameters of the GFP-HUVEC-networks imaged on day 4 of co-culture, analyzed using Angiogenesis Analyzer for ImageJ. The data are represented as mean ± standard deviation. The statistical analysis was performed using GraphPad Prism 9.5.1. An ordinary one-way ANOVA with Dunnett's multiple comparisons test was performed with n ≥ 4; * P < 0.05; ** P < 0.01; *** P < 0.001. (E) Three-dimensional reconstructions generated from confocal stacks (total height: 547.5 µm; z-step: 2.5 µm) of the GFP and F-actin signals for the FGF-2- (top row) and BMP-2- (bottom row) stimulated conditions. Scale bar: 300 µm. Please click here to view a larger version of this figure.
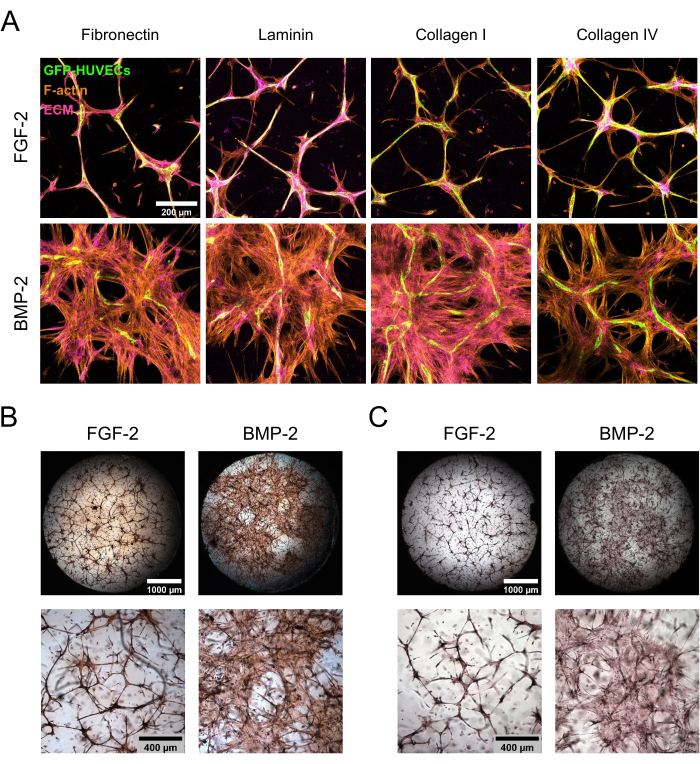
Figure 3: Induction of de-localized hBM-MSC spreading and ECM deposition by BMP-2. (A–C) The 7 day co-cultures grown in the presence of either FGF-2 or BMP-2 were subjected to (A) immunofluorescence or (B,C) direct color staining. (A) The cultures were stained for F-actin and the ECM proteins fibronectin, laminin, collagen I, and collagen IV. The images depict the maximum intensity projections of the confocal stacks (total height: 100 µm; z-step: 5 µm). Scale bar: 200 µm. (B,C) The cultures were stained using (B) Picrosirius Red and (C) Alizarin Red. The top row depicts stitched, whole-well overviews of the images acquired at 2.5x magnification (scale bar: 1,000 µm), while the bottom row depicts one field of view acquired at 5x magnification (scale bar: 400 µm). Please click here to view a larger version of this figure.
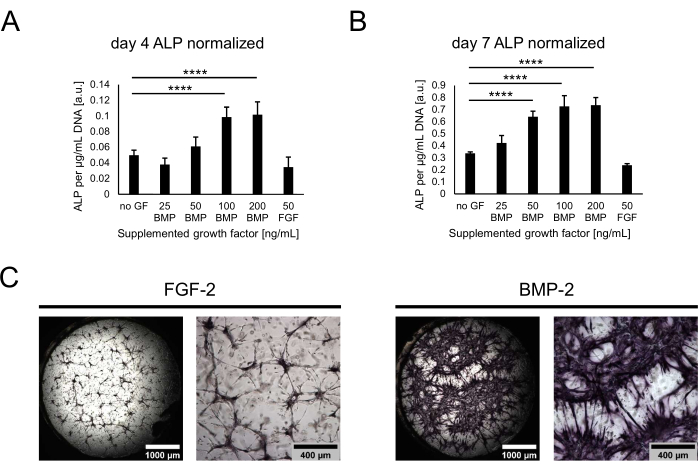
Figure 4: Biochemical assessment of BMP-2-induced ALP activity through direct color staining. (A,B) The ALP activity was determined in the cell lysates of cultures grown in the absence of growth factors or in the presence of BMP-2 at different concentrations or FGF-2 at 50 ng/mL for (A) 4 days or (B) 7 days of co-culture. The ALP activity is shown normalized to the DNA content of each lysate sample. The data are represented as mean ± standard deviation. The statistical analysis was performed using GraphPad Prism 9.5.1. An ordinary one-way ANOVA with Dunnett's multiple comparisons test was performed with n = 5; **** P < 0.0001. (C) Direct color staining of the ALP activity in niches grown in the presence of 50 ng/mL FGF-2 or BMP-2 for 7 days of co-culture. The images on the left-hand side depict stitched whole-well overviews of images acquired at 2.5x magnification (scale bar: 1,000 µm), while the images on the right-hand side depict one field of view acquired at 5x magnification (scale bar: 400 µm). Please click here to view a larger version of this figure.
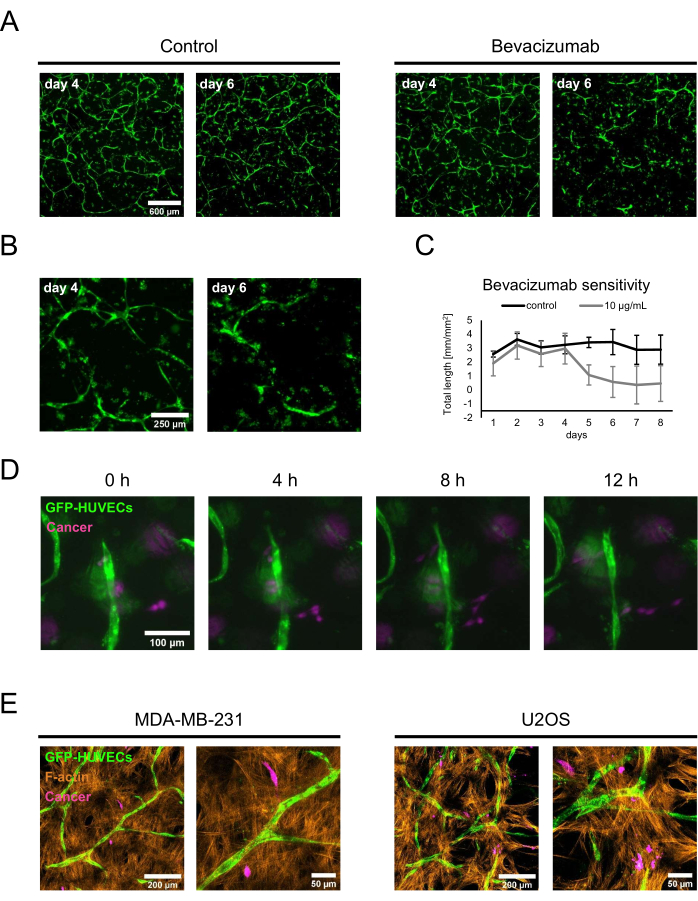
Figure 5: Employment of osteogenic, vascularized niches in advanced cancer models. (A) The GFP signals of cultures grown in the presence of BMP-2 were imaged on day 4 of co-culture before a control solution (left) or bevacizumab at 10 µg/mL (right) were added for 2 days, after which the cultures were imaged again (day 6 of co-culture). Scale bar: 600 µm. (B) Higher magnification images of the bevacizumab-treated cultures are shown in A. Scale bar: 250 µm. (C) The total length of the endothelial networks as shown in A were quantified using Angiogenesis Analyzer for ImageJ from images acquired daily; n ≥ 3. (D) CellTrace FarRed-labeled MDA-MB-231 breast cancer cells were added to the 4 day co-cultures grown in the presence of BMP-2, and time-lapse images were acquired starting 2 days after the cancer cell addition. Scale bar: 100 µm. (E) Maximum intensity projections of confocal stacks (total height: 70 µm; z-step: 2.4 µm) of triple co-cultures generated as described in D and fixed and stained for F-actin. Left: niches featuring MDA-MB-231 breast cancer cells; right: niches featuring U2OS osteosarcoma cells. Scale bars for images on the left: 200 µm; scale bars for images on the right: 50 µm. Please click here to view a larger version of this figure.
