Summary
The pharmacological targeting of ion channels is a promising approach to treating solid tumors. Detailed protocols are provided for characterizing ion channel function in cancer cells and assaying the effects of ion channel modulators on cancer viability.
Abstract
Ion channels are critical for cell development and maintaining cell homeostasis. The perturbation of ion channel function contributes to the development of a broad range of disorders or channelopathies. Cancer cells utilize ion channels to drive their own development, as well as to improve as a tumor and to assimilate in a microenvironment that includes various non-cancerous cells. Furthermore, increases in levels of growth factors and hormones within the tumor microenvironment can result in enhanced ion channel expression, which contributes to cancer cell proliferation and survival. Thus, the pharmacological targeting of ion channels is potentially a promising approach to treating solid malignancies, including primary and metastatic brain cancers. Herein, protocols to characterize the function of ion channels in cancerous cells and approaches to analyze modulators of ion channels to determine their impact on cancer viability are described. These include staining a cell(s) for an ion channel(s), testing the polarized state of mitochondria, establishing ion channel function using electrophysiology, and performing viability assays to assess drug potency.
Introduction
Membrane transport proteins are critical for communication between cells, as well as for maintaining cellular homeostasis. Amongst the membrane transport proteins, ion channels serve to drive the growth and development of cells and to maintain the state of cells in challenging and changing environments. Ion channels have also been reported to drive and support the development of solid tumors, both systemically and in the central nervous system (CNS)1,2. For example, KCa3.1 channels are responsible for regulating membrane potential and controlling cell volume, which is important in cell-cycle regulation. Defective KCa3.1 channels have been reported to contribute to the abnormal proliferation of tumor cells3. Further, ion channels may contribute to the metastatic dissemination of cancers. Transient receptor potential (TRP) channels, for example, are involved in Ca2+ and Mg2+ influx; this influx activates several kinases and heat shock proteins that function to regulate the extracellular matrix surrounding a tumor, which is, in turn, important for initiating cancer metastasis4.
Since ion channels can contribute to the development of cancers, they may also be targets for drug-related cancer treatment. For example, resistance to treatment modalities, including chemotherapy and novel immunotherapy, is related to ion channel function dysregulation5,6,7. In addition, ion channels are emerging as important drug targets to impede the growth and development of cancers, with repurposed small molecule (FDA-approved) drugs being examined, as well as biopolymers, including monoclonal antibodies1,2,8,9. While there has been much progress on this front, ion channel cancer drug discovery remains underdeveloped. This is partly due to the unique challenges of studying ion channels in cancer cells. For example, there are technical limitations in setting up electrophysiology assays for slow-acting compounds and temporal differences in channel activation and drug action. Further, the solubility of compounds can also impede progress, as most of the automated electrophysiology systems commonly in use today utilize hydrophobic substrates, which may contribute to artifacts as a result of compound adsorption. In addition, large bioorganic molecular therapeutics such as natural products, peptides, and monoclonal antibodies are technically challenging to screen using conventional electrophysiology assays10. Finally, the bioelectrical properties of cancer cells remain poorly understood11.
Meanwhile, the immunofluorescence staining of ion channels is often challenging. This is due, in part, to the complexity of their structures and their context in the membrane, which impact the ability to both generate and employ antibodies for microscopy studies. It is especially important that the antibodies used to stain ion channels are validated for specificity, affinity, and reproducibility. Commercial antibodies for ion channels should be considered based on their validation strategy and publication record. Experiments should include negative controls to demonstrate the lack of nonspecific binding by either knockdown or knockout of the target protein. Alternatively, cell lines in which the target protein is absent or in low abundance based on mRNA or protein determinations may serve as negative controls. For example, this study shows the localization of the (GABA) receptor subunit Gabra5 in a medulloblastoma cell line (D283). D283 cells with an siRNA knockdown and Daoy cells, another cerebellar medulloblastoma cell line, were stained for Gabra5 and showed no appreciatable staining (data not shown).
Here, methods are presented to analyze and assay ion channel function, as well as the effect of ion channel modulators on cancer cells. Protocols are provided for (1) staining cells for an ion channel, (2) testing the polarized state of mitochondria, (3) establishing ion channel function using electrophysiology, and (4) in vitro drug validation. These protocols emphasize studies of the type A gamma-aminobutyric acid (GABAA) receptor2,12,13,14,15,16, a chloride anion channel and major inhibitory neurotransmitter receptor. However, the methods presented here apply to studying many other cancer cells and ion channels.
Protocol
1. Immunolabeling ion channels in cultured cells
- Preparing the cells and experimental set-up
- Maintain the cells as an actively growing culture in 75 cm2 culture flasks. Passage the cells once until they become 50%-90% confluent, depending on the doubling time of the cell line being used.
NOTE: For the present study, D283 cells, a Group 3 medulloblastoma cell line, were used. - Collect the cells from the culture flask into a centrifuge tube (15 mL or 50 mL), and add 2 mL of 0.25% trypsin-EDTA to detach adherent cells. Incubate for 5 min at room temperature (RT). After 5 min, tap the flask three to five times to help detach the cells. Collect the cells in 10 mL of medium with 10% FBS to stop the action of the trypsin.
- Centrifuge at 480 x g for 5 min at RT. Gently aspirate the medium. Do not disturb the cell pellet.
- Resuspend the cells in 5 mL to 10 mL of medium, depending upon the density of the culture.
NOTE: The density must be consistent with actively growing cells and is dependent upon the confluency of cells in the flask. Visual assessment should be used to approximate the optimal cell density; specifically, the cells should be between 40%-90% confluent and evenly distributed. - Remove 100 µL of cells, and add to a 1.5 mL tube containing 100 µL of 0.4% trypan blue to mark non-viable cells. Incubate 5-15 min at RT, depending upon the cell line.
- Count the cells manually using a hemocytometer (see the Table of Materials) in 10 µL of solution. Count the live, unstained cells in each of the four corners of the 16 squares of the hemocytometer. Multiply the average cell number with the dilution factor and by 10,000 to give the cells per milliliter (cells/mL). Alternatively, an automated cell counter can be used.
- Dilute cells to 1 x 105 cells/mL into the culture medium specified for each cell line. Seed 1. 8 mL of cells into each well of a 6-well plate containing a 22 mm x 22 mm coverslip pre-coated with 0.1 mg/mL poly-D-lysine as per the manufacturer's instructions (see the Table of Materials).
NOTE: One may want to test the use of both poly-D-Lysine and poly-L-Lysine, as there are reports of cell line-specific preferences17,18. Some cell lines have proteases that can break down poly-L-lysine, while there are reports that poly-D-lysine is toxic to some cell lines. In contrast, neuronal cells, such as PC12, have been reported to be healthier on poly-D-lysine surfaces. - Grow the cells in an incubator with a humidified 5% CO2 environment at 37 °C overnight to allow the cells to attach to the coverslip. Examine the attachment of the cells under an inverted microscope with a 20x objective.
NOTE: The cells must be fully attached to the coverslip before treatment or direct immunostaining. At this time, the cells can be treated with drugs (or vehicle control) by either the addition of a concentrated stock (for example, 600 µL of a 4x stock solution) or the replacement of the medium with fresh medium containing the drug at the desired 1x concentration; the cells can then be returned to the incubator for up to an additional 48 h. Solvent-treated cells are included to assess the effects of the drug on the level and localization of the target molecule. In the present study, D283 cells were treated with 600 µL of a 3.2 µM solution of a GABAA receptor-positive allosteric modulator, QH-II-06614 (see the Table of Materials), for 48 h. The control cells were treated with an equivalent volume of DMSO.
- Maintain the cells as an actively growing culture in 75 cm2 culture flasks. Passage the cells once until they become 50%-90% confluent, depending on the doubling time of the cell line being used.
- Preparation for immunolabeling: Fixation, permeabilization, and blocking
- Rinse the cells with 2.5 mL of 1x PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, see the Table of Materials), and immediately aspirate the solution.
- Fix the cells using 1 mL of 4% PFA in 1x PBS for 1 h at RT.
NOTE: While 4% PFA is the standard fixative for immunostaining, glutaraldehyde or glyoxal can also be used to fix membrane proteins for immunofluorescence. Alcohol-based fixation methods, such as treatments with ice-cold methanol, may lead to less well-preserved morphology and a loss of membrane proteins19,20. - Wash six times with 2.0 mL of 1x PBS (5 min per wash) by agitation on a shaker. Incubate for 1 h at RT in a blocking buffer composed of 1x PBS with 0.8% Triton X-100 and 10% normal serum matching the host species of the primary antibody (alternatively, 3% BSA or bovine serum fraction V albumin can be used in place of normal serum).
NOTE: This step is employed to block non-specific binding.
- Immunolabeling
NOTE: After adding the antibodies conjugated to fluorophores, measurements must be taken soon to prevent photobleaching. Incubations with conjugated antibodies need to be protected from light. The use of a mounting medium that contains agents that scavenge free radicals will reduce photobleaching. Store the slides in a light-tight, opaque container (at 4 °C) after sealing the coverslips. When imaging, photobleaching can be minimized by limiting the intensity of the excitation illumination and the exposure times.- Prepare a humidified chamber by lining the bottom of a 150 mm culture dish with filter paper. Moisten the filter paper with sterile distilled water. Draw a 3 x 2 grid on a piece of laboratory film cut to fit on top of the filter paper, and label it to preserve the orientation of the coverslips in the 6-well plate. Place the laboratory film on top of the filter paper, exposing the upper and lower edges to humidify the chamber.
- Prepare 100 µL per coverslip of the desired dilution of primary antibody in 1x PBS with 3% BSA. To detect the GABRA gene protein product, use rabbit recombinant monoclonal primary antibody at a dilution of 1:200 (Gabra5 antibody, middle region; see the Table of Materials).
NOTE: To empirically determine the primary antibody concentration that produces the optimal signal over background, use a dilution series of the primary antibody while holding the secondary concentration constant. Dilutions of 1:100, 1:250, 1:500, 1:750, and 1:1,000 are recommended to establish optimal primary antibody concentrations. - Transfer the coverslip from the block solution in the 6-well plate to the proper location on the grid drawn on the laboratory film. Discard the block solution from the 6-well plate, and retain the plate for the wash steps.
- Carefully add 100 µL of diluted primary antibody to the center of each coverslip. Avoid placing the solution over the edge of the coverslip. Incubate for 1 hr at RT.
- Add 2.5 mL of 1x PBS to each well of the 6-well plate. Transfer the coverslip back to the appropriate location in the 6-well plate.
- Perform six washes for 5 min each with 2.5 mL of 1x PBS.
NOTE: Do not allow the coverslips to dry during the rinse steps, as this can contribute to a background fluorescence signal. - Return the coverslips to the proper location on the laboratory film. For each coverslip, add 100 µL of secondary antibody diluted in 1x PBS + 1%-5% BSA.
NOTE: Initially, secondary antibodies conjugated to Alexa Fluor 488 (see the Table of Materials) are used at a 1:1,000 dilution in 1x PBS + 3% BSA. The secondary antibody concentration may be optimized by increasing the concentration to detect low-abundance target proteins or decreasing the concentration to reduce the background, if necessary. - For the remaining steps, minimize the light exposure to prevent the photobleaching of the conjugated secondary antibody. Cover the culture dish with aluminum foil, and incubate for 1 h at RT.
- Transfer the coverslips back to the 6-well plate. Using a shaker, perform six washes for 5 min each with 2.5 mL of 1x PBS. Remove the PBS by inverting the plate over the sink or a waste receptacle. After the last wash, leave the PBS in the well to facilitate the removal of the coverslip.
- Label a microscope slide for each coverslip. Add a drop of glycerol-containing mounting medium with DAPI (to stain the nuclei) (see the Table of Materials) to the center of the slide.
- Using forceps, remove the coverslip from the well, and gently place it on the drop of mounting medium on the center of the slide.
NOTE: Avoid the formation of air bubbles in the mounting medium. - Use small pieces of filter paper to remove excess mounting medium. Seal the edges of the coverslip with nail polish, and allow the polish to dry completely before imaging. Store the glass slides at 4 °C in an opaque plastic box.
- Imaging and analysis
- Acquire images with a fluorescence microscope under a 40x objective with immersion oil (see the Table of Materials).
- Visualize the fluorescence images with the appropriate filters.
- NOTE: For Alexa Fluor 488, use an excitation/emission of 496 nm/519 nm; for DAPI, use an excitation/emission of 358 nm/461 nm.
- Capture and save the digital image files. A binning value of 4 x 4 is used to facilitate the image collection rates and reduce the background.
- Prepare the final images. The images can be processed with ImageJ or commercially available software. Results are illustrated in Figure 1.
2. Testing the polarized state of mitochondria
NOTE: This protocol utilizes the TMRE (tetramethylrhodamine, ethyl ester) assay to label the membrane potential in active mitochondria, maintaining a negative charge21,22. TMRE is a cell-permeable, red-orange, positively charged dye accumulating in active mitochondria because of their relative negative charge. Inactive or depolarized mitochondria have reduced membrane potential and fail to proportionally sequester TMRE. FCCP (carbonyl cyanide 4-[trifluoromethoxy] phenylhydrazone), an ionophore uncoupler of oxidative phosphorylation (OXPHOS), depolarizes mitochondrial membranes, thus preventing the accumulation and sequestration of TMRE23. This is illustrated in Figure 2.
- Preparing the cells and experimental set-up
- Maintain the cells in 75 cm2 culture flasks. Aspirate the culture medium, and rinse the cells with 1x PBS without calcium and magnesium.
- Add 2 mL of 0.25% trypsin-EDTA to detach the adherent cells. Incubate for 5 min at RT, and resuspend the cells in 10 mL of medium.
- Collect the cells from the culture flask into a 15 mL centrifuge tube. Centrifuge at 480 x g for 5 min at RT. Aspirate the medium.
- Resuspend the cells in 5 mL to 10 mL of medium, depending upon the original confluency of the culture, so that the cell concentration is 50-100 cells per square on the hemocytometer. Adjust the volume, if necessary, to provide the needed cell density for accurate counting.
- Remove 100 µL of cells, and add to a 1.5 mL tube containing 100 µL of 0.4% trypan blue. Count the cells using a hemocytometer or automated cell counter. Dilute the cells to 3 x 104 cells/mL in culture medium without phenol red.
NOTE: DMEM medium lacking phenol red is used because phenol red exposure inhibits membrane permeability and may contribute to background autofluorescence. - Seed 2.5 mL of the cell suspension (75,000 cells) per well into a 6-well plate containing a 22 x 22 mm coverslip pre-coated with poly-D-lysine as per the manufacturer's instructions (see the Table of Materials).
- Grow the cells in an incubator with humidified 5% CO2 at 37 °C overnight to allow the cells to attach to the coverslip.
- Examine the attachment of cells under an inverted microscope with a 20x objective. The cells must be fully attached to the coverslip before proceeding further.
- Preparing stock solutions
- To prepare a 1 mM stock solution (1,000x) of TMRE (MW = 514.96 g/mol) (see the Table of Materials), dissolve 1 mg of TMRE in 1.94 mL of DMSO. Aliquot and store at −20 °C protected from light. Avoid repeated freeze/thaw cycles.
- To prepare a 50 mM (2,500x) stock solution of FCCP (MW = 254.17 g/mol) (mitochondrial oxidative phosphorylation uncoupler), dissolve 10 mg of FCCP in 786.9 µL of DMSO. Aliquot and store at −20 °C protected from light. Avoid repeated freeze/thaw cycles.
- NOTE: Prepared stock solutions are part of the TMRE Mitochondrial Membrane Potential Assay Kit (see the Table of Materials).
- Establishing the TMRE concentration for the cell lines
- Prepare a 500 nM working TMRE solution in cell culture medium. Protect the working solution from light.
- Add TMRE to cells grown on the coverslips in medium to a final concentration range between 50-200 nM. Do this by first aspirating the culture medium and replacing it with culture medium containing TMRE.
- Incubate for 20 min at 37 °C. Ensure to stagger the treatment times to allow for imaging, since the cells are viewed live.
- Wash the cells twice with 1x PBS, and invert the coverslip on a microscope slide labeled with the final TMRE concentration.
- Immediately image the live cells (peak Ex/Em = 549 nm/575 nm).
NOTE: Immediately image the samples once removed from the culture conditions, since the mitochondrial health is compromised once the cells are removed from the medium and placed on a microscope slide. As an alternative to coverslips, the cells can be imaged in glass-bottom dishes. - Capture the images using the available microscope and software.
NOTE: Establish experimental microscopy imaging parameters during the TMRE concentration optimization process, and maintain the same conditions in subsequent experiments to ensure accurate data comparison. The protocol detailed employed a confocal microscope and compatible software (see the Table of Materials). - Select the optimal TMRE concentration, which is cell line-dependent.
NOTE: The TMRE concentration that provides the best signal to resolve changes in mitochondrial TMRE levels is generally the lowest TMRE concentration, giving an even and readily-detectable fluorescence signal.
- Testing the polarized state of a cell
- Assay preparation
- Prepare the treatment stock solutions at the desired concentrations, and prepare media with the final compound concentration to be assayed. For FCCP, the stock solution should be 20 mM (prepared with DMSO from a 50 mM stock solution).
- Working on a single coverslip at a time, add 1.8 mL of medium plus the test compound to the cells.
- Incubate the cells with the test compound at 37 °C and 5% CO2 in a humidified environment for the desired period of time.
NOTE: The treatment times will vary depending upon the mechanism by which the test compound may alter the mitochondrial membrane potential. Potent protonophores like FCCP that rapidly and directly depolarize mitochondrial membrane potentials require analysis shortly after treatment (within minutes). In contrast, treatment effects with compounds that indirectly impact the mitochondrial electron transport chain function, such as those that alter protein activation or protein turnover or require protein synthesis, may take longer to show an effect. - During incubation, turn on the microscope, and set it to the predetermined optimal imaging parameters, including in terms of the gain, laser intensity, image capture rate, averaging, and exposure time.
- After the drug treatment period, add 200 µL of 10x TMRE (optimal concentration determined above) to the control and drug-treated cells.
- Incubate for 15-30 min at 37 °C in 5% CO2 in a humidified environment. Gently aspirate the medium, and rinse the cells once with 1x PBS.
- Repeat the 1x PBS rinse step to avoid background caused by the cell culture medium or treatment compounds, and invert the coverslip on a microscope slide labeled with the treatment conditions.
- Image the cells live at Ex/Em = 549 nm/575 nm. Using a confocal microscope, collect several z-stack fields with high-speed image capture (512 pixels x 512 pixels, averaging = 4) prior to the loss of cell integrity.
- Save and store the image file for analysis. The procedure is repeated for the next experimental condition.
- Experimental controls
- Use a no-treatment (negative) control, performed as outlined above (steps 3.1.1-3.1.8), lacking the test compound in the medium.
- Use a mitochondrial membrane potential disruption (positive) control consisting of the protonophore compound FCCP. Add 1.8 mL of 20 µM FCCP (see the Table of Materials) in medium to the cells. As described above (steps 3.1.1-3.1.2; and cell imaging, as outlined in steps 3.1.3-3.1.8), incubate for 10 min at 37 °C in 5 % CO2 in a humidified environment prior to staining with TMRE.
NOTE: Both positive and negative controls are required for these experiments.
- Quantification
- Measure the captured pixel intensities of individual cells from a single representative image from the central region of each z-stack. In this work, a single plane of focus was selected to facilitate analysis.
- Analyze the pixel intensities from at least 30 cells (in their entirety) per treatment. Use Excel or Prism to average the pixel intensities for each treatment, and present in bar graph format with the standard error of the mean.
NOTE: ImageJ can also be used for fluorescence quantification. Alternatively, TMRE staining fluorescence can be quantified with commercial software platforms.
- Assay preparation
3. Establishing ion channel function using electrophysiology
NOTE: The procedure in this section describes the use of an automated electrophysiology assay to screen test compounds in a cancer cell line (Figure 3).
- Preparing the cells
- Harvest cells from a healthy culture that lacks dead cells and/or cell overgrowth. Use either the chemical cell detachment solution TrypLE or a manual cell scraper to harvest adherent and clustered cells. Dissociate cells that grow in clumps or spheres, which are particularly common amongst CNS cancer cells.
NOTE: Gentle dissociation of the cells helps maintain the integrity of the membrane proteins essential for ion conductance. - Centrifuge the cells at 561 x g for 10 min at RT. Discard the supernatant, and suspend the pellet in DMEM (without phenol red), HEPES, and penicillin/streptomycin with 20% FBS and 4 mM L-glutamine (see the Table of Materials) maintained at 37 °C.
NOTE: DMEM medium lacking phenol red is used because phenol red exposure inhibits membrane permeability. - Plate the cells on a poly-L-lysine-coated coverslip at low density, so there is separation between the single cells.
NOTE: Poly-L-lysine is used here, and not poly-D-lysine, as poly-L-lysine has better coating properties. - Incubate the cells at 37 °C and 5% CO2 in a humidified chamber for 12 h to 24 h (an optimum time will need to be established) before taking a recording.
- Harvest cells from a healthy culture that lacks dead cells and/or cell overgrowth. Use either the chemical cell detachment solution TrypLE or a manual cell scraper to harvest adherent and clustered cells. Dissociate cells that grow in clumps or spheres, which are particularly common amongst CNS cancer cells.
- Electrophysiology recording
- Remove the medium from the coverslips. Gently wash the cells twice with 1x PBS. Add ~300 µL of TrypLE solution. Using the pipette, gently pipette up/down four to five times until the cells are detached.
- Add serum-free medium (DMEM) (phenol red-free), and maintain a final cell count per volume of at least 1 million cells/mL. Using a 1 mL pipette, declutter the cells to obtain a cell suspension lacking cell clusters.
- Transfer the cell suspension to a 1 mL tube. Incubate the cells at 4 °C for 10 min to stabilize the cell membrane. Keep the cells rotating on a shaker (gentle cycle) to avoid the formation of cell clumps.
- Prepare the Port-a-Patch setup (see the Table of Materials). Turn on the computer, amplifier, and perfusion system. Open the Patch-Master software, and create a new file.
- Bring the buffers (extracellular and internal) to RT, and then load the buffers into the respective perfusion tubes.
NOTE: Recordings of the GABAA receptor require both an "extracellular (bath) solution" containing 161 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM HEPES, and 6 mM D-glucose (pH adjusted to 7.4 using NaOH) and an "internal solution" containing 120 mM KCl, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES (pH adjusted to 7.2 using NaOH). - Fill the bottom of an electrode chip (manufacturer-provided) with 5 µL of internal solution, and place the electrode chip on the Faraday top of the Port-a-Patch. Add 10 µL of external solution to the chip, and observe the rectangular pulse on the oscilloscope using the Patch-Master software (see the Table of Materials).
NOTE: The resistance may be in the range of 2-3 MΩ, but this is cell line-dependent. - Start recording once the rectangular pulse is visible, after the initial electronic adjustments, and after the software prompts the user to add cells.
- Gently add 20 µL of single-cell suspension onto the central part of the electrode, and wait for cell patch followed by a Giga-Ohm seal by observing the resistance values in the software.
- The live progress of cell patching can be observed on the left column of the software. Once the cell is "maintained in whole-cell mode", begin the recordings by opening a protocol within the software.
- Set the holding potential at −80 mV. Run a gap-free continuous recording protocol using the Patch-Master software to record the current from the cell.
- Obtain a stable baseline, and switch to a ligand and respective compound application for a specified duration by using the automated perfusion tab on the left panel of the software.
- Acquire data using the Patch-Master software.
NOTE: The data are low-pass filtered at 1 kHz and digitalized at 100 kHz. - Perform data analysis by computing the maximum amplitude of the current response using the Nest-o-Patch software (see the Table of Materials).
4. In vitro potency
NOTE: This procedure details a MTS assay to determine drug potency. The One Solution Cell Proliferation Assay combines all the required assay reagents into a prepared solution that can be added in one step to cell culture wells to assess the cell viability and proliferation after treatment with experimental compounds. The reagent is reconstituted as per the manufacturer's recommendations (see the Table of Materials), aliquoted, and stored at −20 °C. The section describes the use of the assay to determine the IC50 of test compounds in a particular cell line (Figure 4). This MTS reagent can also be used for the high-throughput screening of large numbers of compounds at known concentrations.
- Preparing the cells
NOTE: The cell number depends upon the growth and metabolism of each individual cell line. Determine the optimal cell number experimentally prior to using the assay to evaluate any investigational treatments.- Harvest adherent cells from the culture by trypsinization, and use Accutase (see the Table of Materials) to dissociate the cells that grow in clumps or spheres.
NOTE: Glioblastoma and primary medulloblastoma cell lines often require Accutase as they tend to grow in clumps or spheres. - Count the cells, and make a cell solution with 10,000 cells per well in a 75 µL volume. Make dilutions of the stock for the cell numbers to be tested. Prepare at least 1 mL per dilution.
- Plate 75 µL of each dilution into sets of five consecutive wells in a 96-well plate. Incubate at 37 °C and 5% CO2 overnight in a humidified environment (i.e., an incubator).
NOTE: Cell lines with slow growth or metabolism (that acidify their growth medium in more the 3 days) may require more than 10,000 cells as the upper cell number range, while cell lines that grow rapidly (generally faster than 20 h doubling time) require fewer cells. Cell lines with a high metabolism may require fewer than 1,000 cells per well. The cell range can be adjusted to determine the optimal plating density for the MTS assay for these cell lines.
- Harvest adherent cells from the culture by trypsinization, and use Accutase (see the Table of Materials) to dissociate the cells that grow in clumps or spheres.
- Mock treatment control
- Add 25 µL of medium per well to simulate the addition of the test compound in the MTS assay.
NOTE: In the actual MTS assay, 25 µL of a 4x stock of the chosen treatment concentration for the test compound (here a GABAA receptor modulator, QH-II-066) will be added to the cells to give the 1x final treatment concentration in a total volume of 100 µL per well. - Incubate at 37 °C and 5 % CO2 in a humidified environment for 48 h. This simulates standard incubation in the presence of a drug.
- Add 25 µL of medium per well to simulate the addition of the test compound in the MTS assay.
- MTS assay
- Thaw the required aliquots of the MTS reagent. Add 20 µL of MTS reagent per well. Incubate at 37 °C and 5 % CO2 in a humidified environment for 1 h.
- Centrifuge the plate (1,350 x g for 5 min) at room temperature to remove bubbles, which can interfere with the absorbance reading.
NOTE: Remove any bubbles with a fine-gauge needle. - Read the absorbance at 490 nm. Save the data as an Excel file.
- Return the plates to 37 °C in 5% CO2, and read repeatedly within the 4 h linear range period of the assay.
NOTE: There is no need to re-centrifuge before reading the absorbance. Data from later readings, especially the 2 h reading, may be important for examining how fast a cell line metabolizes the MTS reagent and are useful in selecting the number of cells to the plate for the assay. - Plot the absorbance optical density (OD) at 490 nm (OD490) versus the plated cell number using Excel or Prism.
- Select the cell number for the assay. The optimal number of cells will be in the linear range and below saturation (OD490 = 1).
NOTE: For cells with slow growth, the highest cell number plated can be adjusted to 20,000 cells per well, and 500 cells per well can be left out of the assay.
- Determining the IC50
- Day 1: Plate the cells for the MTS assay
- Record the cell line name and passage number of each line in the study. To counter evaporation during the assay, at a minimum, fill the outer perimeter top and bottom rows and the end left and right perimeter columns of the 96-well plate with 1x PBS, 100 µL per well, to counter evaporation during the assay.
- Plate the optimal number of cells for each cell line as determined prior to the start of the assay. Plate the cells in 75 µL per well of phenol red-free, antibiotic-free medium that does not contain HEPES buffer. The FBS can be increased to 20% to support the growth of the cell lines.
NOTE: Medium lacking phenol red is used because phenol red exposure inhibits membrane permeability. - Plate the sample in at least quintuplicate for treatment concentration. Count the cells using a manual or automated hemocytometer.
NOTE: To use a manual hemocytometer, (1) prepare a 1:1 dilution of cells resuspended in medium in trypan blue; (2) incubate 5-15 min, depending upon the cell line; (3) load 10 µL of the cells in trypan blue into the hemocytometer counting chamber; (4) count the viable cells in the four corners and middle squares, and determine the average number of cells per square; (5) calculate the viable cells per milliliter as follows:
Viable cells per mL = Average viable cells × 2 (dilution factor) × 104 - Use a sufficient volume to prepare the desired concentration of cells in phenol red-free, antibiotic-free medium that does not contain HEPES buffer (i.e., 75 µL per well x 60 wells per plate = 4.5 mL of cells per plate).
NOTE: Each plate needs to have a medium-only control for the background subtraction. The medium control can replace the PBS wells if necessary.
- Day 2: Adding the treatment compound
- Prepare the experiments treatments at 4x the desired final concentration to give the desired final concentration when 25 µL of treatment solutions are added to cells plated in 75 µL of medium (= 100 µL total volume per well).
- Prepare the test solutions (in this case, the GABAA receptor modulator QH-II-066) by serial dilution of the highest-concentration stocks. For example, if the final drug concentrations are to be 5.0 µM, 2.5 µM, 1.25 µM, 0.625 µM, and 0.3125 µM, then prepare 1:1 serial dilutions beginning with 20 µM to obtain 10 µM, 5 µM, 2.5 µM, and 1.25 µM concentrations, respectively.
NOTE: Each plate requires two cell-based controls in addition to the medium-only control: untreated cells (medium alone) and cells solvent-treated (often DMSO) at a known concentration. It is good practice to ensure that the vehicle does not affect cell proliferation in the active range of the compound. - Once the test compound is added, incubate the cells at 37 °C and 5% CO2 in a humidified environment for 48 h.
- Day 3: Performing the cell proliferation assay (MTS)
- Calculate the volume of MTS reagent needed (20 µL per 100 µL of testing culture), and thaw the required volume of aliquoted MTS reagent (see the Table of Materials).
- Vortex, and ensure that the reagent is completely in solution before use. Pipet the thawed reagent into a sterile 25 mL reagent reservoir.
- Pipet 20 µL of MTS reagent (using a multichannel pipette) into each well of the 96-well plate containing samples in 100 µL of culture medium.
- Incubate the plate at 37 °C and 5% CO2 in a humidified environment for 1 h to 4 h. Plates can be read at multiple time points. Generally, the plates are read at 1 h and at 2 h or 3 h.
- Bubbles in the medium can lead to erroneous results. Prior to reading in the plate reader, spin the plate at 1,350 x g for 5 min at room temperature. A needle or a pipette tip can be used to remove any bubbles that remain after centrifugation.
- Measure the absorbance at 490 nm using a suitable microplate reader or spectrophotometer (see the Table of Materials).
- Save the data as an Excel file.
NOTE: The incubation time in the presence of the test compound will vary depending upon the rate of metabolism and the growth of the cell line under study, in addition to the compound being tested. An incubation period of 48 h is a good starting point for most cell lines and test compounds.
- Day 1: Plate the cells for the MTS assay
- Data analysis
- Transfer the data in an Excel file, and organize the data for each replicate by increasing test compound concentration. Average the medium-only control value, and subtract this value from the experimental data.
- Determine the percent of cell proliferation in the treatment wells compared to the vehicle wells (or solvent-treated control wells) by setting the control to 100% proliferation for each replicate.
- Transfer the data generated in Excel into a program of choice. The authors generally use Prism software (see the Table of Materials) to determine the drug concentration that inhibits the cell proliferation by 50% (IC50).
- In Prism software, create a data table with X-column as cell numbers and Y-column as the average OD for five replicate values in side-by-side sub-columns.
- Enter the treatment concentration values in the X column when analyzing a drug treatment. Label the x-axis with the treatment compound name and concentration units.
- Enter the calculated replicate cell viability values from the Excel analysis into the Y sub-columns. Label the y-axis as cell proliferation (%).
- Using the analysis tool, select Non-linear regression (curve fit) under XY analyses.
- Select dose-response [inhibition] vs. normalized response– variable slope.
- Perform the analysis function. View the results table to see the calculated best-fit values for IC50 and the curve fit graph. The x-axis of the graph can be changed to a log scale if desired.
Representative Results
Above are select procedures that can be employed to characterize ion channels in cancerous cells. The first protocol highlights the staining of an ion channel. As detailed, there are many challenges when staining an ion channel or, for that matter, any protein that is present in the extracellular membrane. Shown in Figure 1 is the staining for a subunit of the pentameric GABAA receptor. The second protocol highlights the results of testing the polarized state of mitochondria in cancerous cells. Mitochondria play roles essential for cell viability and proliferation, as well as cell death. In mammalian cells, mitochondria activate apoptosis in response to cellular stress through the release of the Bcl-2 family proteins found in between the mitochondrial inner and outer membranes. In the cytosol, the Bcl-2 family proteins activate caspase proteases, which mediate programmed cell death. Alterations in the plasma membrane ion channel function can result in a disruption of intracellular ion homeostasis, including in terms of the ion levels within the mitochondria, which can lead to a loss of membrane potential, thus triggering apoptosis14. Levels of Ca2+, K+, Na+, and H+ are important determinants in signaling events that can trigger mitochondrial-initiated cell death. Shown in Figure 2 is staining with the cell-permeable, positively charged dye TMRE to label and image the membrane potential in active mitochondria, which maintain a negative charge21,22. TMRE is a red-orange dye that binds with active mitochondria because of their relative negative charge. Depolarized or inactive mitochondria have reduced membrane potential and, thus, fail to sequester TMRE. In this experiment, the ionophore uncoupler FCCP is an important control, as it depolarizes mitochondrial membranes, thereby preventing the accumulation of TMRE23. The third protocol highlights single-cell patch-clamp electrophysiology. Shown in Figure 3 are representative recordings of a trace recorded from the patient-derived medulloblastoma cell line D283. Finally, the fourth protocol highlights an assay to determine the state of proliferation of cancerous cells. Shown in Figure 4 are details on how the MTS assay works and an illustration of the plate and readout when incubated with an agent that impairs the viability of the cancer cells under study (in this case, DAOY).
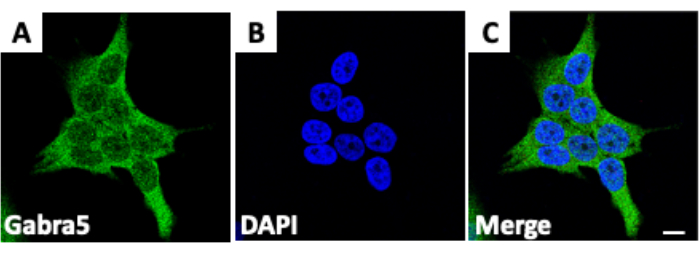
Figure 1: Staining cells for ion channels. (A) Staining of the protein Gabra5, a subunit of the GABAA receptor, in D283 medulloblastoma cancer cells. (B) Fixed cells treated with the fluorescent stain 4′,6-diamidino-2-phenylindole (DAPI), which binds to DNA. (C) Merge of medulloblastoma cancer cells stained for both Gabra5 and DAPI. Scale bars = 10 µm. The figure is adapted from Kallay et al.14. Please click here to view a larger version of this figure.
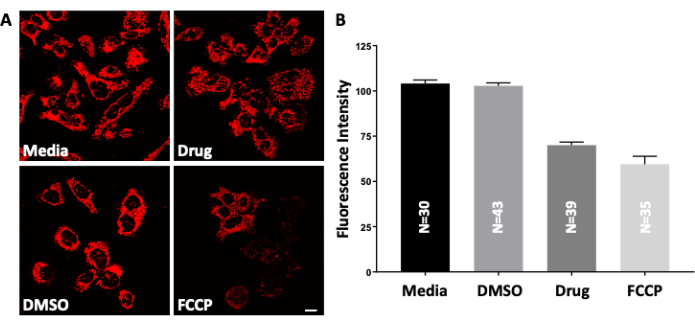
Figure 2: Testing the polarized state of mitochondria. (A) Live D283 medulloblastoma cancer cells are treated with increasing concentrations of the drug QH-II-066. The cells are then treated with positively charged, cell-permeable TMRE (tetramethylrhodamine, ethyl ester), which accumulates in active (negatively charged) mitochondria. Depolarized or inactive mitochondria have reduced membrane potential and, therefore, fail to retain the TMRE dye; as a result, they show a low fluorescence signal. Imaged by fluorescence microscopy; FCCP (carbonyl cyanide 4-[trifluoromethoxy] phenylhydrazone). Peak: λex, 549 nm; λem, 575 nm. Scale bars = 10 µm. (B) Quantification of TMRE staining (images shown in panel A) using the software platform. Data are presented as the mean and standard error of the mean. The figure is adapted from Kallay et al.14. Please click here to view a larger version of this figure.
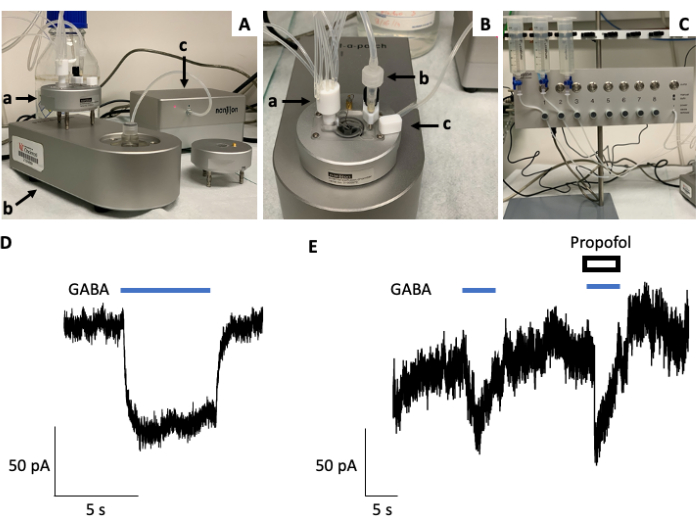
Figure 3: Establishing ion channel function using electrophysiology. (A) Shown is a Port-a-Patch setup or rig, consisting of a Faraday cage (a), recording chamber (b), and suction unit (c, right). (B) Top view of the Port-a-Patch rig highlighting the recording chamber equipped with a perfusion inlet (a), outlet (b), and reference electrode (c). (C) The Port-a-Patch is connected to a rapid solution exchange perfusion system with automated and manual modes of operation and solution reservoirs. (D) Representative current trace from a whole-cell patch-clamp electrophysiology recording using a Port-a-Patch rig (Nanion) and D283 medulloblastoma cancer cells. GABA (10 µM) was applied for 5 s with a holding potential of −80 mV. (E) Representative current trace from a whole-cell patch-clamp electrophysiology recording using a Port-a-Patch rig (Nanion) and D283 medulloblastoma cancer cells with the co-application of GABA (1 µM) and the GABAA receptor agonist (general anesthetic) propofol (50 µM), which potentiates the current induced by GABA alone. Please click here to view a larger version of this figure.
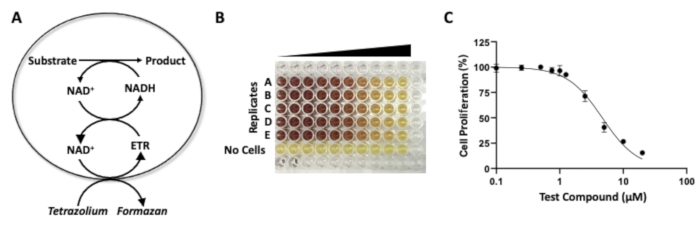
Figure 4: MTS assay to assess drug potency. (A) Chemical reactions underlying the "MTS assay" used to assess the potency of an agent, as reflected by a reduction in cell proliferation. Reduction of MTS tetrazolium by cells that are viable, generating the dye formazan. (B) A 96-well plate showing the colorimetric results of an MTS assay with increasing drug concentrations. In this experiment, the DAOY medulloblastoma cancer cells are treated with increasing concentrations of a pre-clinical drug, KRM-II-08, a positive allosteric modulator of the GABAA receptor. (C) A dose-response curve generated with the MTS assay (from the quantification of 96-well plate). Please click here to view a larger version of this figure.
| Manual | Semi/fully automated | |
| Through-put | Low | High |
| Fast solution exchange | Possible | Yes |
| Cost | High | Low |
| Operator | Experienced | Beginner/Intermediate |
| Cell type | All cells; tissues | Primary single cells; cell lines |
| Cell numbers required | Minimum* | High* |
| Drug/Solution volume | High | Low |
| Resources/Utilities | High | Low |
| Maintenance | High | Low |
| Experiment control | Very good | Good |
| Live cell imaging | Yes | No |
| *Port-a-Patch, for example, requires at least a million cells/mL for recordings, while a manual set-up usually needs a few hundred cells on a coverslip. | ||
Table 1: Comparison of manual versus semi and/or fully automated electrophysiology setups.
Discussion
Changes in ion channel function alter intracellular signaling cascades, which can impact the overall functioning of a cell. Over the past decade, it has become increasingly clear that ion channels are important to cancer cell growth and metastasis. Importantly, many ion channels are primary targets for approved therapeutics targeting a broad range of disorders24. Investigators have probed whether ion channels could be anti-cancer targets, and the initial results are promising2,16,25. The field is just beginning to investigate the role of ion channels in cancer development and as therapeutic targets, and the future looks bright on both fronts.
In this work, detailed procedures are provided for analyzing ion channels in cancer cells and determining whether a channel is a therapeutic vulnerability. These assays serve as a guide to aid in studying ion channels in cancer cells. Methods have been described that focus on the visualization of ion channels in cancerous cells, the determination of how the modulation of ion channels alters the polarized state of cancerous cells, different approaches to analyze ion channel function using electrophysiology, and the measurement of cancer cell viability.
Immunofluorescence staining can be used to detect the presence and cellular localization of ion channels. The experimental conditions must be carefully optimized to accurately represent where a channel is found within the cell. Intuitively, one would expect the staining of ion channels to be relatively straightforward, as they are most likely solvent accessible. However, it is important to remember that the immunofluorescence epitope may not be easily accessible, depending on whether it is part of an extracellular or intracellular domain. Furthermore, ion channels are often densely clustered on cell membranes, so their detection by immunostaining may require more extensive optimization of the fixation and permeabilization procedures compared to other classes of proteins14,26.
The mitochondrial membrane potential is essential for many mitochondria-associated processes, including ATP synthesis, the generation of reactive oxygen species (ROS), calcium sequestration, the import of mitochondrial proteins, membrane dynamics, and triggering apoptosis. This protocol utilizes the TMRE assay to label membrane potential in active mitochondria in single cells. For high-throughput screens, the TMRE levels can be measured with a fluorescence plate in a microplate format.
Patch-clamp electrophysiology is the “gold standard” method for the study of ion channel kinetics. Whole-cell and single-channel methods are also the highest-resolution methods for accurately determining the structure-function relationships and pharmacology of ion channel modulators. This method utilizes the study of small electrical changes across a membrane caused by the movement of ions through ion channels27. Electrophysiology experiments are traditionally performed using manual patch-clamp electrophysiology recordings of a single cell at a time, resulting in a low-throughput approach that requires the user to possess a series of specialized skill sets. Automated patch-clamp electrophysiology techniques, such as Port-a-Patch, IonFlux Mercury, Patchliner, and/or Synchropatch technologies, offer multiple recordings at a time, and these techniques are equipped with a sophisticated perfusion setup for compound application, resulting in semi-high throughput or high throughput without requiring special skills. For some experiments, manual patch clamping is irreplaceable (Table 1). Still, automated patch-clamp technology has accelerated ion channel biophysics research and related drug discovery programs. One of the limitations of recording from primary/non-transfected cells is the contribution of leaky, non-specific endogenous currents. In our recordings, we observed slight baseline variations, suggesting a potential contribution of endogenous currents from D283 cells.
Cell proliferation assays measure the perturbation of cell growth activity in response to a chemical agent. Such assays are critical tools to assess the action of a drug on cell proliferation. To evaluate cell proliferation, investigators most commonly utilize MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), MTS (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium), and/or clonogenic assays. MTT and MTS assays measure the conversion (reduction) of a water-soluble tetrazolium salt (yellow) into a quantifiable formazan product (purple in the MTT assay), which is catalyzed by the dehydrogenase enzyme system of metabolically active cells. The intensity of the colored product provides an estimation of the number of “viable” (e.g., metabolically active) cells. In contrast, the clonogenic assay analyzes the ability of single cells in culture to form a colony (at least 50 cells or six successive divisions) following treatment with a drug28,29. MTT, MTS, and clonogenic assays each have advantages and disadvantages, and one should carefully assess the suitability of these assays to determine the potency of a drug(s) with a given cell line(s). Optimally, using more than one of these assays (e.g., clonogenic and MTT or MTS assays) is recommended to determine the potency of a drug(s).
The MTT and MTS assays assess the metabolic activity of living cells, which can vary by cell type and assay condition. The MTT or MTS assays are advantageous, as these assays are easy to perform, can be performed in replicate, and are amenable to high-throughput screening30. The MTT or MTS assays can be completed in 3-4 days, while the clonogenic assay can take 10-21 days, depending on the characteristics of the cell line used28. In contrast, the disadvantages of both the MTS and MTT assays are possible interference of the reagents required for performing the assay with culture medium, the difficulty of using the assay, and the potential variability between replicates due to the cell metabolic status and culture conditions31. Regarding the choice between the MTT and MTS assays, one should consider that the MTT assay requires a solubilization step to lyse the cells and allow the formazan crystals to dissolve in the medium and shows absorbance at 570 nm, whereas the MTS assay is a “one-step assay”, in which the formazan is directly solubilized into the medium without the intermittent steps (e.g., the cell lysis step). The formazan product in the MTS assay is darker in color and possesses a more sensitive absorbance value range (490-500 nm), thus making it easier to ascertain a positive response. The MTS assay is faster than the MTT assay, as 2-3 h is needed for the reaction versus 4 h for the reaction plus 1-2 h solubilization in the MTT assay. Additionally, the MTS assay formazan product remains in suspension, and the assay is more suitable for suspension cells than the MTT assay, since no medium change is required during the MTS assay32. However, a modified MTT assay has been developed in which the solubilization step has been improved using a combination of DMSO and SDS lysis solution. This modified MTT assay can be used for both adherent and suspension cells. If one opts to use the MTT assay, then care should be taken in selecting the cell culture and the conditions/reagents used for cell growth. For example, the results will vary if the cells are grown as a monolayer, differentiated, as a confluent monolayer, or senescent. In addition, certain non-mitochondrial dehydrogenases, like some intracellular reductases and flavin oxidases, can reduce the MTT reagent, possibly eliciting false-positive readings33. Further, the MTT reagent has a degree of toxicity, so the incubation time should be limited. The rate of MTT reduction can also change with the culture conditions, such as the pH and glucose content of the medium and the physiological state of the cells32,34. For example, the presence of ascorbic acid reportedly reduces MTT to formazan, and this effect is enhanced in the presence of retinol35. Regarding potential issues with the clonogenic assay, it is important to be aware that following drug addition, the cells can become prematurely senescent and “clonogenically inactive” (i.e., they do not form colonies) but remain metabolically active and exhibit activity in the MTT or MTS assays. Another point to remember is that the clonogenic assay is limited to the study of adherent cells, and not all adherent cells can form colonies when plated at low cell densities, since cell-cell communication is lost36. Due to inadequate cell-cell communication and the limitations of self-produced growth factors, the clonogenic assay responds to lower drug doses than the MTT and MTS assays.
Offenlegungen
The authors have nothing to disclose.
Acknowledgements
The authors acknowledge support from the Thomas E. & Pamela M. Mischell Family Foundation to S.S. and the Harold C. Schott Foundation funding of the Harold C. Schott Endowed Chair, UC College of Medicine, to S.S.
Materials
| ABS SpectraMax Plate Reader | Molecular Devices | ABS | |
| Accutase | Invitrogen | 00-4555-56 | |
| Alexa Flor 488 | Invitrogen | A32723 | Goat Anti-Rabbit |
| Antibiotic-Antimycotic | Gibco | 15240-062 | 100x |
| B27 Supplement | Gibco | 12587-010 | Lacks vitamin A |
| Biosafety Cabinet | LABCONCO | 302381101 | Class II, Type A2 |
| Bovine Serum Albumin | Fisher Scientific | BP1606-100 | |
| CO2 Incubator | Fisher Scientific | 13-998-211 | Heracell VIOS 160i |
| Calcium Chloride | Fisher Scientific | C7902 | Dihydrate |
| Cell Culture Dishes, 150 mm | Fisher Scientific | 12-600-004 | Cell culture treated |
| Cell Culture Flasks, 75 cm2 | Fisher Scientific | 430641U | Cell culture treated |
| Cell Culture Plates, 6 well | Fisher Scientific | 353046 | Cell culture treated |
| Cell Culture Plates, 96 well | Fisher Scientific | 353072 | Cell culture treated |
| Centrifuge | Eppendorf | EP-5804R | Refrigerated |
| Corning CoolCell | Fisher Scientific | 07-210-0006 | |
| Coverslips, 22 x 22 mm | Fisher Scientific | 12-553-450 | Corning brand |
| D283 Med | ATCC | HTB-185 | |
| DABCO Mounting Media | EMS | 17989-97 | |
| D-Glucose | Sigma Life Sciences | D9434 | |
| Dimethyl Sulfoxide | Sigma Aldrich | D2650 | Cell culture grade |
| DMEM/F12, base media | Fisher Scientific | 11330-032 | With phenol red |
| DMEM/F12, phenol red free | Fisher Scientific | 21041-025 | |
| EGTA | Sigma Aldrich | E4378 | |
| Epidermal Growth Factor | STEMCELL | 78006.1 | |
| FCCP | Abcam | AB120081 | |
| Fetal Bovine Serum, Qualified | Gibco | 10437-028 | |
| Fibroblast Growth Factor, Basic | Millipore | GF003 | |
| GARBA5 Antibody | Aviva | ARP30687_P050 | Rabbit Polyclonal |
| Glutamax | Gibco | 35050-061 | |
| Glycerol Mounting Medium | EMS | 17989-60 | With DAPI+DABCO |
| Hemocytometer | Millipore Sigma | ||
| Heparin | STEMCELL | 7980 | |
| HEPES | HyClone | SH3023701 | Solution |
| HEPES | Fisher Scientific | BP310-500 | Solid |
| ImageJ | Open platform | With Fiji plugins | |
| Immuno Mount DAPI | EMS | 17989-97 | |
| KRM-II-08 | experimental compounds not available from a commercial source | ||
| Leica Application Suite X | Leica Microsystems | ||
| Leukemia Inhibitory Factor | Novus | N276314100U | |
| L-Glutamine | Gibco | 25030-081 | |
| Magnesium Chloride | Sigma Aldrich | M9272 | Hexahydrate |
| Microscope, Confocal | Leica | SP8 | |
| Microscope, Light | VWR | 76382-982 | DMiL Inverted |
| MTS – Promega One Step | Promega | G3581 | |
| Multi-channel pipette, 0.5-10 µL | Eppendorf | Z683914 | |
| Multi-channel pipette, 10-100 µL | Eppendorf | Z683930 | |
| Multi-channel pipette, 30-300 µL | Eppendorf | Z683957 | |
| Nest-O-Patch | Heka | ||
| Neurobasal-A Medium | Gibco | 10888022 | Without vitamin A |
| Neurobasal-A Medium | Gibco | 12348-017 | Phenol red free |
| Non-Essential Amino Acids | Gibco | 11140-050 | |
| NOR-QH-II-66 | experimental compounds not available from a commercial source | ||
| Parafilm | Fisher Scientific | 50-998-944 | 4 inch width |
| Paraformaldehyde | EMS | RT-15710 | |
| PATHCHMASTER | Heka | ||
| Penicillin-Streptomycin | Gibco | 15140-122 | |
| Perfusion System | Nanion | 4000120 | |
| PFA | EMS | RT-15710 | |
| Phosphate Bufered Saline | Fisher Scientific | AAJ75889K2 | Reagent grade |
| Poly-D-Lysine | Fisher Scientific | A3890401 | |
| Poly-L-Lysine | Sigma Life Sciences | P4707 | |
| Port-a-Patch | Nanion | 21000072 | |
| Potassium Chloride | Sigma Life Sciences | P5405 | |
| Primary Antibody | Invitrogen | MA5-34653 | Rabbit Monoclonal |
| Prism | GraphPad | ||
| Propofol | Fisher Scientific | NC0758676 | 1 mL ampule |
| QH-II-66 | experimental compounds not available from a commercial source | ||
| Reagent Reservoirs | VWR | 89094-664 | Sterile |
| Slides, 75 x 25 mm | Fisher Scientific | 12-544-7 | Frosted one side |
| Sodium Bicarbonate | Corning | 25-035-Cl | |
| Sodium Chloride | Fisher Scientific | S271-3 | |
| Sodium Pyruvate | Gibco | 11360-070 | |
| Synth-a-Freeze Medium | Gibco | R00550 | Cryopreservation |
| TMRE | Fisher Scientific | 50-196-4741 | Reagent |
| TMRE Kit | Abcam | AB113852 | Kit |
| Triton X-100 | Sigma Aldrich | NC0704309 | |
| Trypan Blue | Gibco | 15-250-061 | Solution, 0.4% |
| Trypsin/EDTA | Gibco | 25200-072 | Solution, 0.25% |
| Vortex Mixer | VWR | 97043-562 | |
| Whatman Filter Paper | Fisher Scientific | 09-927-841 |
Referenzen
- Prevarskaya, N., Skryma, R., Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies. Physiological Reviews. 98 (2), 559-621 (2018).
- Rao, R., et al. Ligand-gated neurotransmitter receptors as targets for treatment and management of cancers. Frontiers in Physiology. 13, 839437 (2022).
- Mohr, C. J., et al. Cancer-associated intermediate conductance Ca2+-activated K+ channel KCa3.1. Cancers. 11 (1), 109 (2019).
- Fels, B., Bulk, E., Petho, Z., Schwab, A. The role of TRP channels in the metastatic cascade. Pharmaceuticals. 11 (2), 48 (2018).
- Eil, R., et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 537 (7621), 539-543 (2016).
- Haustrate, A., Hantute-Ghesquier, A., Prevarskaya, N., Lehen’kyi, V. Monoclonal antibodies targeting ion channels and their therapeutic potential. Frontiers in Pharmacology. 10, 606 (2019).
- Kischel, P., et al. Ion channels: New actors playing in chemotherapeutic resistance. Cancers. 11 (3), 376 (2019).
- Tuszynski, J., Tilli, T. M., Levin, M. Ion channel and neurotransmitter modulators as electroceutical approaches to the control of cancer. Current Pharmaceutical Design. 23 (32), 4827-4841 (2017).
- Kale, V. P., Amin, S. G., Pandey, M. K. Targeting ion channels for cancer therapy by repurposing the approved drugs. Biochimica Biophysica Acta. 1848 (10), 2747-2755 (2015).
- Wickenden, A., Priest, B., Erdemli, G. Ion channel drug discovery: Challenges and future directions. Future Medicinal Chemistry. 4 (5), 661-679 (2012).
- Rocha, P. R. F., Elghajiji, A., Tosh, D. Ultrasensitive system for electrophysiology of cancer cell populations: A review. Bioelectricity. 1 (3), 131-138 (2019).
- Sengupta, S., et al. α5-GABAA receptors negatively regulate MYC-amplified medulloblastoma growth. Acta Neuropathologica. 127 (4), 593-603 (2014).
- Jonas, O., et al. First in vivo testing of compounds targeting Group 3 medulloblastomas using an implantable microdevice as a new paradigm for drug development. Journal of Biomedical Nanotechnology. 12 (6), 1297-1302 (2016).
- Kallay, L., et al. Modulating native GABAA receptors in medulloblastoma with positive allosteric benzodiazepine-derivatives induces cell death. Journal of Neurooncology. 142 (3), 411-422 (2019).
- Pomeranz Krummel, D. A., et al. Melanoma cell intrinsic GABAA receptor enhancement potentiates radiation and immune checkpoint response by promoting direct and T cell-mediated anti-tumor activity. International Journal of Radiation Oncology, Biology, Physics. 109 (4), P1040-P1053 (2021).
- Bhattacharya, D., et al. Therapeutically leveraging GABAA receptors in cancer. Experimental Biology and Medicine. 246 (19), 2128-2135 (2021).
- Mazia, D., Schatten, G., Sale, W. Adhesion of cells to surfaces coated with polylysine. Applications to electron microscopy. Journal of Cell Biology. 66 (1), 198-200 (1975).
- Wiatrak, B., Kubis-Kubiak, A., Piwowar, A., Barg, E. PC12 cell line: Cell types, coating of culture vessels, differentiation and other culture conditions. Cells. 9 (4), 958 (2020).
- Baker, J. R. Fixation in cytochemistry and electron-microscopy. Journal of Histochemistry and Cytochemistry. 6 (5), 303-308 (1958).
- Chung, J. Y., et al. Histomorphological and molecular assessments of the fixation times comparing formalin and ethanol-based fixatives. Journal of Histochemistry and Cytochemistry. 66 (2), 121-135 (2018).
- Crowley, L. C., Christensen, M. E., Waterhouse, N. J. Measuring mitochondrial transmembrane potential by TMRE staining. Cold Spring Harbor Protocols. 2016 (12), (2016).
- Cory, A. H., Owen, T. C., Barltrop, J. A., Cory, J. G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Communications. 3 (7), 207-212 (1991).
- Maro, B., Marty, M. C., Bornens, M. In vivo and in vitro effects of the mitochondrial uncoupler FCCP on microtubules. EMBO Journal. 1 (11), 1347-1352 (1982).
- Zheng, J., et al. Mechanism for regulation of melanoma cell death via activation of thermo-TRPV4 and TRPV. Journal of Oncology. 2019, 7362875 (2019).
- Konno, K., Watanabe, M., Luján, R., Ciruela, F. Immunohistochemistry for ion channels and their interacting molecules: Tips for improving antibody accessibility. Receptor and Ion Channel Detection in the Brain. , (2016).
- Mortensen, M., Smart, T. G. Single-channel recording of ligand-gated ion channels. Nature Protocols. 2 (11), 2826-2841 (2007).
- Franken, N. A., Rodermond, H. M., Stap, J., Haveman, J., van Bree, C. Clonogenic assay of cells in vitro. Nature Protocols. 1 (5), 2315-2319 (2006).
- Rafehi, H., et al. Clonogenic assay: Adherent cells. Journal of Visualized Experiments. (49), 2573 (2011).
- Scudiero, D. A., et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Krebsforschung. 48 (17), 4827-4833 (1988).
- Wang, P., Henning, S. M., Heber, D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS One. 5, e10202 (2010).
- Berridge, M. V., Tan, A. S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Archives of Biochemistry and Biophysics. 303 (2), 474-482 (1993).
- Plumb, J. A., Milroy, R., Kaye, S. B. Effects of the pH dependence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide-formazan absorption on chemosensitivity determined by a novel tetrazolium-based assay. Krebsforschung. 49 (16), 4435-4440 (1989).
- Chakrabarti, R., Kundu, S., Kumar, S., Chakrabarti, R. Vitamin A as an enzyme that catalyzes the reduction of MTT to formazan by vitamin C. Journal of Cellular Biochemistry. 80 (1), 133-138 (2000).
- Dong, G. W., Preisler, H. D., Priore, R. Potential limitations of in vitro clonogenic drug sensitivity assays. Cancer Chemotherapy and Pharmacology. 13 (3), 206-210 (1984).
- Sun, J., et al. STIM1- and Orai1-Mediated Ca2+oscillation orchestrates invadopodium formation and melanoma invasion. Journal of Cell Biology. 207 (4), 535-548 (2014).
