The crystallization facility and VMXi beamline have been used for a wide variety of project types and use cases. Here are a small number of examples to illustrate what users may wish to pursue.
Case study 1: Standard data collection
The beamline enables rapid determination of room-temperature crystal structures from a small number of crystals within a crystallization plate. The minimum number of crystals is dependent on the space group and crystal orientations but is frequently 1-4 although improved data quality may be achieved by merging data from several tens of crystals. A recent example is one of the beamline standards, thaumatin. Multiple crystals, shown in Figure 8A, were marked up for data collection manually as described in protocol section 2.3. These crystals were added to the queue as described in protocol section 2.4 and experimental parameters were selected from the dropdown list. Once experimental parameters had been applied, the plate was queued for data collection. Datasets were collected, automatically scaled, and merged using the xia2.multiplex pipeline as described in protocol section 3. An example output from SynchWeb is shown in Figure 8A middle. Five merged datasets gave rise to a dataset of 1.66 Å resolution. For standard data collection of approximately five crystals in a well, datasets were collected within 2.5 min.
Case study 2: Ligand Binding – Fragment experiment using Mac1 protein
Producing structures of protein-ligand complexes at room temperature may be straightforwardly achieved using the beamline. Ligands may be added to drops on crystallization plates (either manually or by acoustic drop injection) and data measured after a suitable incubation time. In the example described here, a series of fragments were dispensed into wells containing crystals of the SARS-CoV-2 first macrodomain of the protein nsp3 (Mac-1) in a crystallization plate. Two of the wells containing the same fragment were assigned as a group as described in protocol step 2.5. Multiple crystals (42) were marked for data collection as described in protocol steps 2.3 and 2.4, and datasets were collected using standard parameters (60° rotation, 0.1° step, 0.00178 s exposure, 5% transmission, 16 KeV – per crystal) (Figure 8B). Datasets from the two wells were automatically processed using the xia2.dials pipeline and subsequently, the xia2.multiplex pipeline was initiated to automatically merge 22 of these data sets. DIMPLE was then run on the output of these pipelines and yielded maps that clearly showed evidence of the bound fragment. The fragment model was built into the unoccupied density and further refined (Figure 8B right). Room-temperature ligand-bound structures can easily be determined using this series of steps to provide invaluable information and feedback to the structure-based drug design process. For this data collection of 42 crystals across a number of wells, datasets were collected within 10 min.
Case study 3: Structure solution with a low symmetry space group and preferred orientations A stack of multiple crystals with plate-like morphology was produced from crystallization experiments with a c-type gas-binding cytochrome (Figure 8C). By selecting several positions around the edge of the stack where only a single crystal was in the X-ray beam, it was possible to obtain a good quality dataset to 1.75 Å resolution by merging wedges from four crystals, despite a monoclinic (C2) space group. This allowed rapid progress of the project without the need to further optimize the crystallization conditions. This result has been described previously9. For this data collection of four crystals in a well, datasets were collected within 2 min.
Case study 4: Obtaining information and room-temperature structure from microcrystals in a plate using serial crystallography
Often when microcrystals appear in a drop or when users are seeking to optimize micro-crystallization protocols in batch as a precursor to serial crystallography experiments at synchrotron or XFEL sources, it is very helpful to gain rapid feedback on the diffraction properties and unit cell dimensions of different trials using minimal material. In this use case, microcrystals of lysozyme growing in batch were pipetted into a crystallization plate (200 nL volume per drop) and data collected from eight drops using a grid scan with a 10 µm step size (Figure 9). The resulting 25,906 still images were processed using serial crystallography software resulting in a dataset, where 9,891 diffraction patterns were indexed and merged producing a dataset to 2.0 Å resolution that refined well against the published room temperature structure (Rwork = 19.6%, Rfree = 23.6% using PDB 8A9D) (Table 1). This allowed detailed analysis of unit cell distribution and a microcrystal room temperature structure determination that could feed into complex serial crystallography experiments including time-resolved studies. The total volume of microcrystal suspension required was 1.6 µL. For this data collection of microcrystals across eight wells using grid scans, datasets were collected within 40 min.
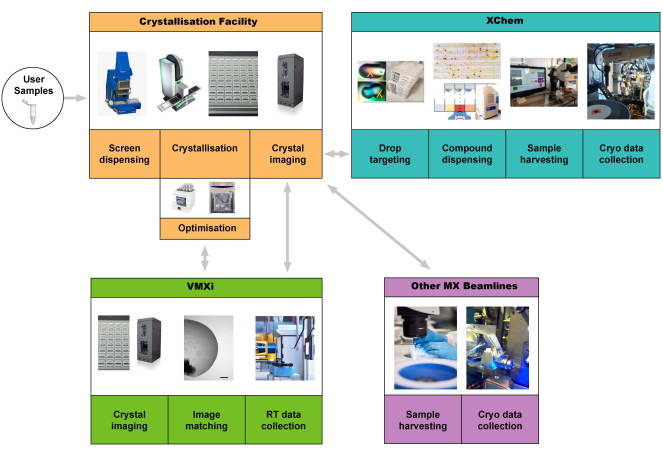
Figure 1: Schematic of the protein-to-structure pipeline integrating crystallization screening, optimization at the crystallization facility, room-temperature automated data collection and processing without sample harvesting at VMXi, XChem fragment screening, and data collection at other MX beamlines. Users can start the pipeline by supplying a sample or by bringing plates to the VMXi beamline. Abbreviation: Versatile Macromolecular Crystallography in situ. Please click here to view a larger version of this figure.

Figure 2: The Mosquito SPT Labtech interface for setting up crystallization plates. (A) (1) The MiTeGen In Situ-1 Setup view. Choose the MiTeGen 2 drop standard plate by going to (2) the 96-well plate type and selecting (3) the MiTeGen plate 2 drop plate. To change the definition parameters for drop 1 and drop 2, which is required for VMXi, click on the (4) edit icon. This opens a new window (B) where (5) the X and Y offsets need to be changed as shown. Select (B) the sub well 2 and (C) sub well 3 and change the values accordingly. (D) The CrystalQuickX Setup view. Choose the CrystalQuickX 2 drop standard plate by going to the 96-well plate type and selecting the MiTeGen plate 2 drop plate. To change the definition parameters for drop 1 and drop 2, which is required for VMXi, click the edit icon the same as above. This opens a new window where (E,F) the X and Y offsets need to be changed as shown. Please click here to view a larger version of this figure.
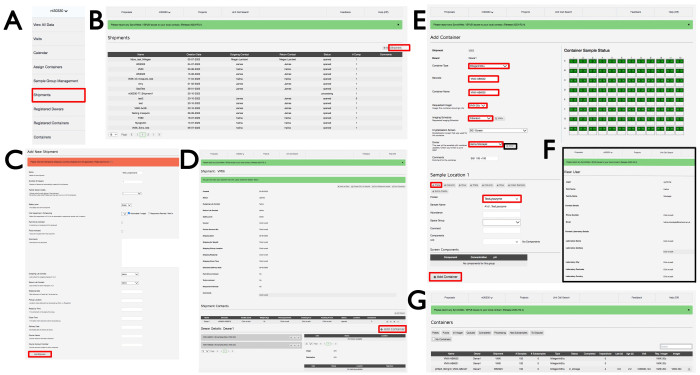
Figure 3: The SynchWeb interface showing how to create a VMXi shipment, register a plate, and check contact details. Screenshots of the various stages of uploading information into the SynchWeb interface are shown from (A) the dropdown menu, (B,C) registering a new shipment, (D) registering a new container, (E) inputting the plate information, (F) check contact details, and (G) a list of registered containers within a proposal. Please click here to view a larger version of this figure.
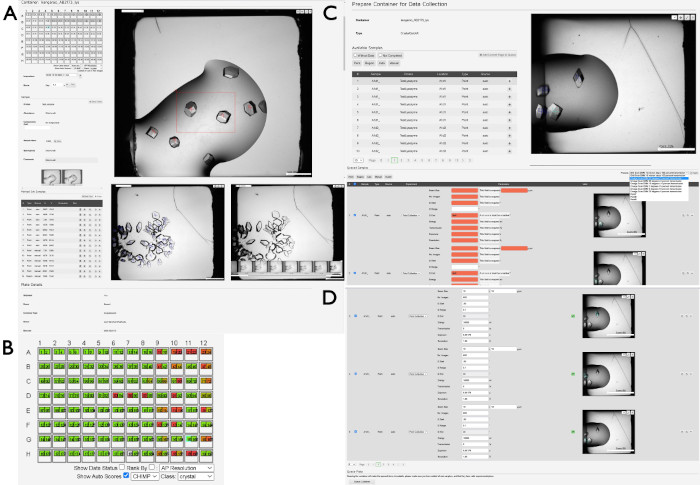
Figure 4: Selecting and preparing samples for data collection using SynchWeb. A series of screenshots showing the various stages of preparing samples for data collection using the SynchWeb interface are displayed. (A) Points and regions of interest are selected from the drop overview. In the lower section of this panel, there is a chronological series of photographs of one drop. (B) An example of the CHiMP output for one plate highlighting results for the 'crystal' category. (C) Adding samples to the queue from the list of selected points and regions and (D) applying parameters for data collection to the queued samples from the dropdown list of beamline-created experiment settings. Note the difference between samples with no experimental parameters (in red) versus those that have correctly applied parameters (top and bottom). At the bottom of this panel is the Queue Container button, which queues the plate to be collected on the beamline. Please click here to view a larger version of this figure.
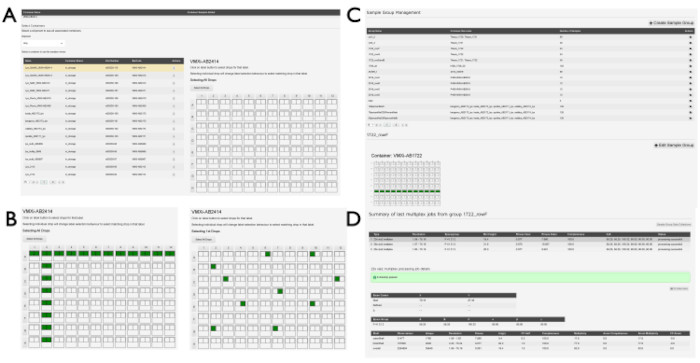
Figure 5: Sample group creation in SynchWeb. A series of screenshots showing the various stages of creating sample groups. (A) The plate(s) containing samples are selected from the relevant shipment and (B) the drops within the plate are selected. These may be individual drops or may be selected by row and/or column. (C) A list of sample groups that have already been created. (D) The outputs of the last three multiplex processing jobs are listed and can be selected to show statistics from the processing pipeline. Please click here to view a larger version of this figure.
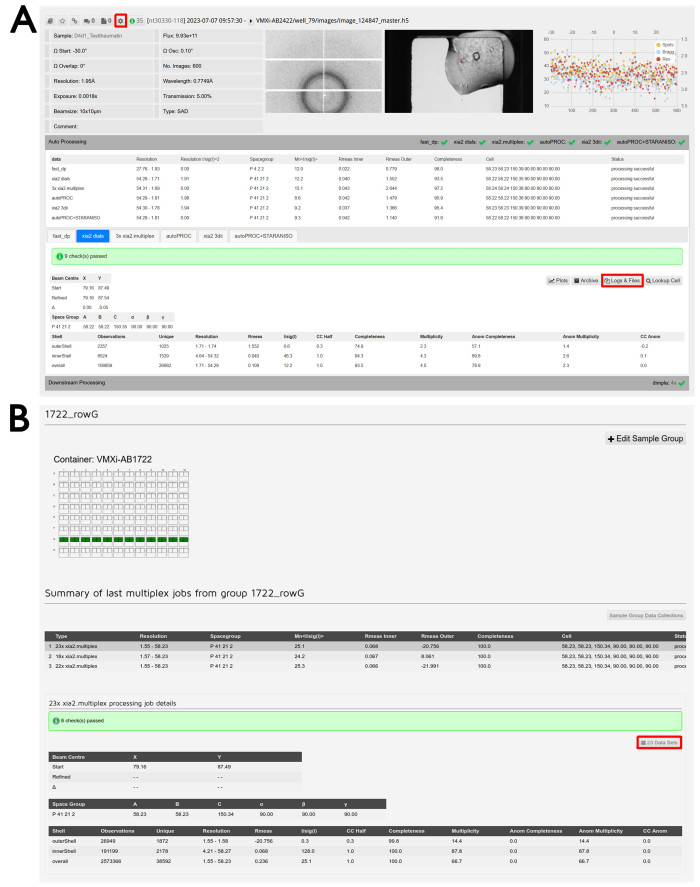
Figure 6: Data processing and data reduction. (A) Screenshot of a processed dataset in ISPyB11. The button to access reprocessing features is highlighted. Sample ID and experimental parameters are shown in the top left and the diffraction image viewer in the middle. Clicking on this image will open an interactive window to examine different images. The crystal image viewer is shown on the right and clicking on this image will also open an interactive window to compare beamline and Formulatrix storage images. Per-image analysis plot is shown on the far right and clicking on this image will open a magnified version of this output. Clicking on the Auto Processing tab will make the autoprocessing visible and make the comparison between the results of the different pipelines easy. Click on the tabs to switch between the different processing pipelines and view the detailed output from the selected pipeline. The Logs & Files button for data download is highlighted. Clicking on the Downstream Processing tab will expand and provide results for any data sets run through post-data reduction pipelines where appropriate. (B) Screenshot from the Sample Group Management screen. The user-defined group name is on the top and the visual description of the included wells can be seen below. A green well indicates that all crystals measured from that drop will be included in the group. A summary of different multiplex jobs performed on that group can be seen and underneath is the detailed output from multiplex. The Data Sets button to examine the included experiments is highlighted. Please click here to view a larger version of this figure.
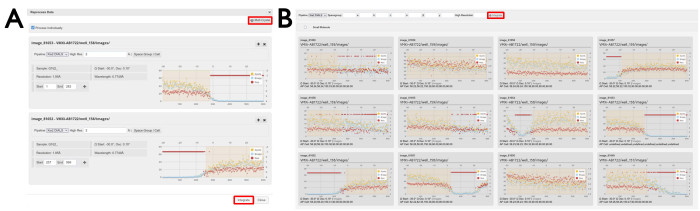
Figure 7: Data reprocessing windows. (A) Individual and (B) multi-crystal datasets. Two individual datasets are displayed where regions of data have been selected. With the Process individually check box ticked, the selected diffraction images will be individually processed by pressing the Integrate button. Clicking the Multi-crystal button will open a display of the individual datasets. To reprocess diffraction images from multiple datasets, regions of images are selected as displayed, and reprocessing is initiated by clicking the Integrate button as highlighted. Please click here to view a larger version of this figure.

Figure 8: Representative results from the VMXi pipeline. (A) Marked-up crystals for protein thaumatin within a crystallization drop (left panel), data processing results (center panel), and electron density (right panel). (B) Collection on multiple crystals to determine binding of the fragment to SARS-CoV-2 Macro domain. Datasets were collected on multiple crystals in the presence of a fragment from the EU-OPENSCREEN fragment screen using standard experimental settings. Examples of these data collections are shown in this excerpt from SynchWeb. The fragment was built into the corresponding density and further refined as is displayed in the furthest on the right. (C) Marked-up monoclinic crystals in a stack from a challenging crystallization hit used for data collection. Green crosses and red numbers indicate where data were measured using a 10 µm beam and 60° rotation. Four of the resulting wedges were merged to produce a dataset at 1.75 Å resolution. Electron density around the Heme group is displayed on the right. Please click here to view a larger version of this figure.

Figure 9: Serial crystallography in the crystallization plate. (A) Optical image of the crystallization drop, with a white box representing the region of interest. (B) Definition of grid scan points. (C) Heat map indicating diffraction. (D) Electron density map arising from a serial crystallography data set from greater than 9,000 still diffraction patterns. Please click here to view a larger version of this figure.
| Resolution (Å) | Completeness (%) | Multiplicity | I/σ(I) | Rsplit | CC1/2 | Unique Observations |
| Overall | 100 | 95.5 | 20.8 | 0.063 | 0.998 | 8422 |
| Low (55.55 – 5.43) | 100 | 147.1 | 81.7 | 0.028 | 0.999 | 488 |
| High (2.03 -2.00) | 100 | 75.3 | 1.2 | 1.092 | 0.410 | 411 |
Table 1: Data statistics for the VMXi RT serial dataset. Abbreviations: I = mean intensity of the scaled observations; Rsplit = a measure of discrepancy of the measured intensities; CC 1/2 = correlation coefficient between two random halves of the dataset.
