The outcomes of the 1,536-well crystal screening experiment consist of seven complete brightfield image sets collected at day 0 (negative control), day 1, week 1, week 2, week 3, week 4, and week 6 (Figure 4). SONICC images are collected at the 4 week time point for plates incubated at 23 °C and at the 6 week time point for plates incubated at 4 °C or 14 °C. Altogether, once a sample has been shipped, users can anticipate having their plates set up within 1 day of arrival. The images will be uploaded as they are collected. The crystallization screening experiment concludes after 6 weeks.
The 1,536-well plate setup allows all the screening experiments to be conducted within the same plate, thus limiting sample consumption and facilitating imaging and direct comparison between imaging modalities. Representative results for the time course of crystal growth for a single cocktail condition are shown in Figure 4. Automated plate imaging throughout the course of the experiment allows the identification of both rapidly and slowly growing crystals by brightfield imaging. The UV-TPEF and SHG imaging allow cross-validation of the hits observed by brightfield imaging and indicate that the crystals observed are proteinaceous and crystalline, respectively (Figure 5A,B). Furthermore, SONICC imaging enables the identification of crystals that are visually obscured by precipitate or films (Figure 5C) or microcrystals that may otherwise be mistaken for precipitate (Figure 5D). For some crystals, a lack of SHG signal is not disqualifying, as some point groups do not produce an SHG signal35,36, as exemplified by the tetragonal thaumatin crystal in Figure 5C. Conversely, a lack of UV-TPEF signal for proteins lacking tryptophan residues should be anticipated. The observation of UV-TPEF and SHG signals also facilitates the identification of non-protein salt crystals, which will appear in brightfield and exhibit a strong positive SHG signal but will lack a UV-TPEF signal (Figure 5E).
Image analysis for the plate setup is streamlined with the MARCO Polo GUI, which also bundles the ftp data transfer from the HWI servers (as an alternative to transferring files with FileZilla). The MARCO Polo GUI allows for easily navigable plate and image viewing and performs computational image scoring using the MARCO algorithm so that the image results can be rapidly downloaded, viewed, and analyzed from the HTX Center. The MARCO scoring algorithm, as implemented in the MARCO Polo GUI, is capable of scoring images from the entire 1,536-well plate in less than 5 min. Images flagged as crystalline by the MARCO algorithm can be subsequently sorted by the Polo GUI for display. Since the MARCO algorithm was optimized for crystal identification and minimizing false negatives so as not to miss any positive hits, the scoring can result in false positive flags. Nevertheless, the ability of MARCO to limit the set of images needing to be examined by focusing attention on the wells with a high probability of containing crystals results in a substantial reduction in data processing burden for users. The convenient implementation of the algorithm in the user-friendly MARCO Polo viewing platform, with its ability to sort images based on MARCO scores, greatly improves the user's ability to analyze the dataset quickly and to accurately determine crystal hits.
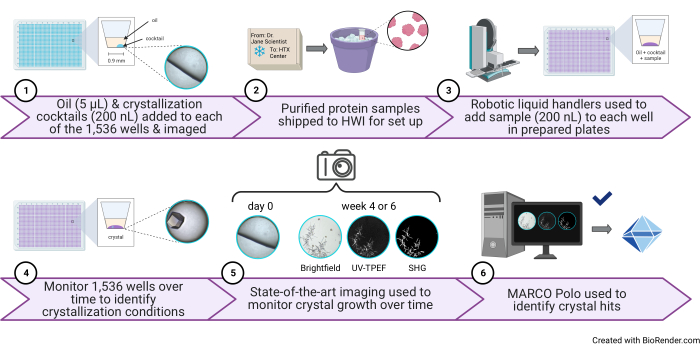
Figure 1: Schematic of a high-throughput 1,536-well crystallization screening experiment performed at the HTX Center. (1) In this step, 5 µL of paraffin oil and 200 nL of cocktail are added to each well (protocol step 3.1 and step 3.5). A cartoon illustration of one well containing only oil and cocktail and a representative image are shown to the right. (2) Samples arrive at the HTX Center (protocol step 5.1). 3) In this step, 200 nL of sample is added to each well (protocol step 5.4). (4) All 1,536 wells are monitored over time using brightfield imaging, 5) as well as the UV-TPEF and SHG modalities (protocol step 6). 6) The AI-enabled open-source GUI is used to view, score, and analyze the crystallization images (protocol step 7). Abbreviations: HTX = high-throughput crystallization; UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation; AI = artificial intelligence; GUI = graphical user interface. Please click here to view a larger version of this figure.

Figure 2: Single 1,536-well plates containing screening experiments, imaged using brightfield, UV-TPEF, and SHG imaging. The 1,536-well plates are shown with an American penny for scale (top). Each screening experiment is imaged once prior to setup and six times after sample addition with brightfield imaging (seven total brightfield image sets, left). The plates undergo UV-TPEF (center) and SHG (right) imaging at 4 weeks or 6 weeks. Abbreviations: UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation. Please click here to view a larger version of this figure.
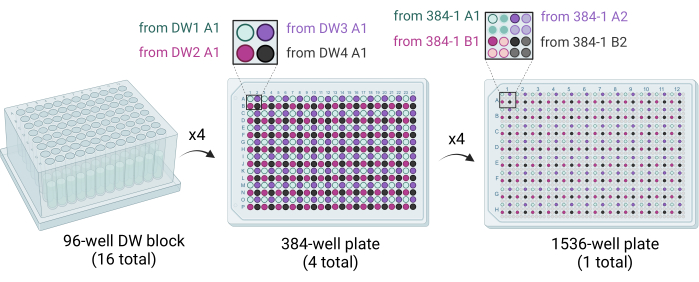
Figure 3: Schematic showing how the 1,536-well plates are generated. Sixteen 96-well DW blocks are used to stamp out four 384-well plates, with each quadrant of each 384-well plate filled by dispensing crystallization cocktails. Four 96-well DW blocks fill one 384-well plate (middle). Four 384-well plates are used to stamp out the single 1,536-well plate (right). Abbreviation: DW = deep well. Please click here to view a larger version of this figure.
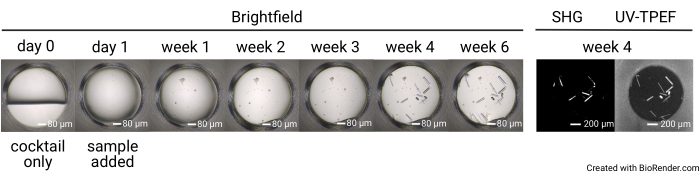
Figure 4: Representative time course of a single well in a 1,536-well screening experiment. Plates are imaged prior to sample setup (day 0), as well as with brightfield imaging on day 1, week 1, week 2, week 3, week 4, and week 6. The plates incubated at 23 °C are imaged with SONICC at week 4. Scale bars = 80 µm (brightfield), 200 µm (SHG, UV-TPEF). Abbreviations: SONICC = second order nonlinear imaging of chiral crystals; UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation. Please click here to view a larger version of this figure.
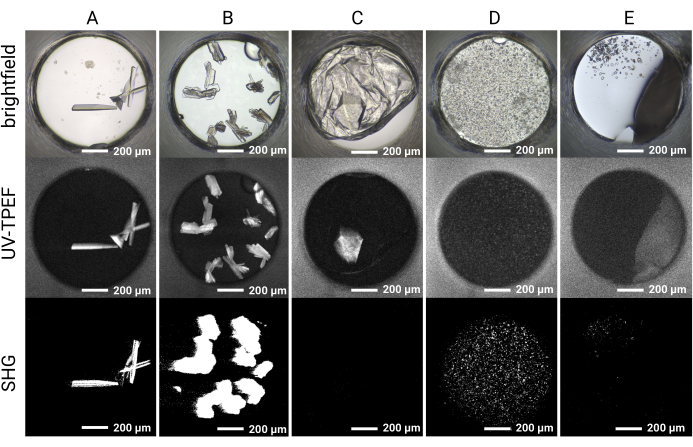
Figure 5: Representative imaging results for the HT 1,536 crystal screening experiments. Brightfield, UV-TPEF, and SHG imaging results are shown for five example wells. (A,B) Protein crystals observed by brightfield, UV-TPEF, and SHG imaging are clearly apparent in all three imaging modalities. (C) A protein crystal obscured by film in brightfield imaging is visible by UV-TPEF imaging; the crystal is not observed by SHG imaging due to point group incompatibility. (D) Example of microcrystals verified by UV-TPEF and SHG imaging that may otherwise be considered precipitate. (E) Example of salt crystals that appear crystalline by brightfield and SHG imaging but do not exhibit a UV-TPEF signal. Scale bars = 200 µm. Well diameter = 0.9 mm. Abbreviations: UV-TPEF = UV-two-photon excited fluorescence; SHG = second harmonic generation. Please click here to view a larger version of this figure.
Supplementary Figure S1: Opening image files in MARCO Polo. Image files can be opened within the MARCO Polo GUI by navigating to the Import | Images tab at the top (a). Note that files can also be transferred via the From FTP tool directly in MARCO Polo (a) or can be transferred via FileZilla as described in protocol step 7.2. To import files that have already been downloaded, select Images | From Rar Archive/Directory. In the popup window that appears, select Browse for Folder (b), and navigate to the file directory where the plate image files are saved. Once the files are in the Selected Paths window (c), highlight a file, and click on Import Runs (d). The MARCO Polo GUI will identify the correct Cocktail File metadata to import with the images. Please click here to download this File.
