1. Rearing and selecting Drosophila larvae
- Maintain/culture the fly strain (w1118, wildtype) on standard fly food medium at 25 ˚C in a humidified incubator.
- Select 10 wandering 3rd instar larvae 72 h after egg laying.
- Place the larvae in a 35 mm empty Petri dish and gently wash them by transferring the larvae to the new dish containing tap water using forceps. Do this 2x to remove any remaining food.
2. Brain isolation from Drosophila larvae (Figure 1)
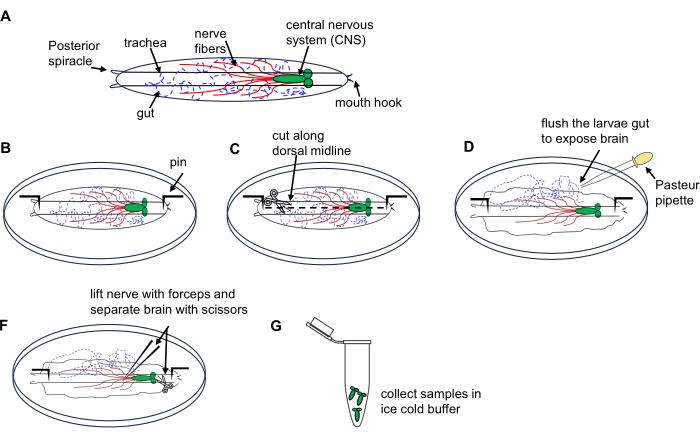
Figure 1: Dissection of Drosophila larval brain from 3rd instar wandering stage. (A) Schematic drawings of Drosophila larva. (B–G) Larva dissection. Please click here to view a larger version of this figure.
- Place 10 larvae on a dissection dish containing ice-cold PBS.
- Place the larva dorsal side up (identified by tracheal tubes running along its length) and pin each end to the bottom of the dish followed by making a small incision on the body wall at the posterior end.
- Cut the body wall along the dorsal midline towards the anterior end using microdissection scissors.
- Briefly flush the interior of the larva using a Pasteur pipette 3x with PBS in the dish to expose the brain.
- Locate and lift the brain using the forceps and carefully isolate it using microdissection scissors.
- Transfer the dissected brains to a 1.5 mL microcentrifuge tube filled with ice-cold PBS on ice; collect all larva brains. Proceed with RNA extraction using an RNA microprep kit as described in section 4.
NOTE: Do a dissection of 10 larvae within 15 min to prevent tissue damage and RNA degradation.
3. Drosophila S2 Schneider cells
- Grow Drosophila S2 cells in Drosophila Schneider's medium supplemented with 10% fetal bovine serum (FBS) at 25 °C in a humidified incubator to a density of 8 × 106 to 10 × 106 cells/mL with minimum 90% viability.
- In a 50 mL sterile conical tube, dilute cells to 2.5 × 106 cells/mL with Schneider's Drosophila medium supplemented with 10% FBS that has been prewarmed to 25 °C.
- Transfer 8 mL of the cell suspension (20 × 106 cells) into a 100 mm culture plate and add 4 mL of medium to make it to 12 mL (day 1).
- Incubate the cultured cells at 25 °C in a humidified incubator.
NOTE: Cells loosely adhere to the plate after 12-16 h (day 2). - Transfect the cells with appropriate DNA plasmids24.
- Incubate for 48 h in a humidified incubator.
- After incubation, collect cells by adding 5 mL of ice-cold PBS by gentle pipetting (day 4).
- Transfer the cells to a 15 mL tube.
- Pellet the cells by centrifuging at 1,000 × g for 5 min at 4 °C.
- Rinse the cells 2x with ice-cold PBS by gentle pipetting and collect the cells by centrifuging them at 1,000 × g for 5 min at 4 °C.
- Perform RNA extraction using an RNA miniprep kit.
NOTE: Perform the following steps inside a sterile laminar flow hood.
4. Total RNA extraction from Drosophila larvae brain and S2 cells
- Larval brain: Remove PBS by brief centrifugation (8 s short spin at 5,000 × g).
- Add 600 µL of RNA lysis buffer and homogenize 10x with a plastic pestle. Visually inspect the tube under a stereo microscope to ensure complete lysis.
- Centrifuge at 1,000 × g for 5 min at 4 °C to remove tissue debris. Transfer the cleared supernatant into a nuclease-free microcentrifuge tube.
- Isolate RNA using an RNA microprep kit according to the manufacturer's instructions.
NOTE: The use of an RNA microprep kit is essential for the larval brain samples because of the small amount of RNA present in the samples. - S2 cells: Remove PBS and isolate RNA according to the manufacturer's instructions.
- Measure RNA yield and quality by spectrophotometry and agarose gel electrophoresis.
- Determine the purity and quantity of extracted RNA by measuring the optical density of the extracted RNA at A260 nm and A280 nm, respectively. Make sure the A260 nm/A280 nm ratio is ≥2.0 and the RNA concentration is >350 ng/µL for downstream applications.
NOTE: A typical RNA yield from 10 Drosophila larva brains is ~500-800 ng/µL or 2.5-4 µg in 5 µL. For S2 cells, the yield is ~2-3 µg/µL (15-30 µg in 15 µL). Isolated RNA can be stored at -80 °C for long-term storage.
- Determine the purity and quantity of extracted RNA by measuring the optical density of the extracted RNA at A260 nm and A280 nm, respectively. Make sure the A260 nm/A280 nm ratio is ≥2.0 and the RNA concentration is >350 ng/µL for downstream applications.
5. Preparation of RNA gel and electrophoresis
- 1.5% Denaturing RNA gel (100 mL)
NOTE: Formaldehyde is toxic through skin contact and inhalation of vapors; handle it in a chemical fume hood.- Dissolve three agarose tablets (1.5 g) in 82 mL of MOPS buffer (Supplemental File 1) until tablets completely break up to form fine particles.
- Heat the agarose slurry in a microwave until the solution is clear and all particles are completely dissolved.
- Cool the solution to ~60 °C.
- Add 18 mL of 37% formaldehyde, then mix by gentle swirling. Pour the solution into the casting tray and let it solidify in a fume hood.
- RNA sample preparation and electrophoresis
- Dilute the RNA sample to 200 ng (in 5 µL) and add 5 µL of 2x RNA loading dye.
- Heat the samples at 70 ˚C for 5 min in a dry bath.
- Load 2 µL of RNA ladder in the first lane and 10 µL of samples in the adjacent lanes.
- Perform electrophoresis in MOPS buffer at 100 V for 60 min for a 5 x 6 cm gel.
NOTE: Adjust the electrophoresis conditions depending on the amplicon sizes. - Visualize the gel on a UV transilluminator.
NOTE: The presence of a single band ~600 nucleotide size indicates intact RNA preparation (see Figure 2A).
6. Poly(A) tail length measurement
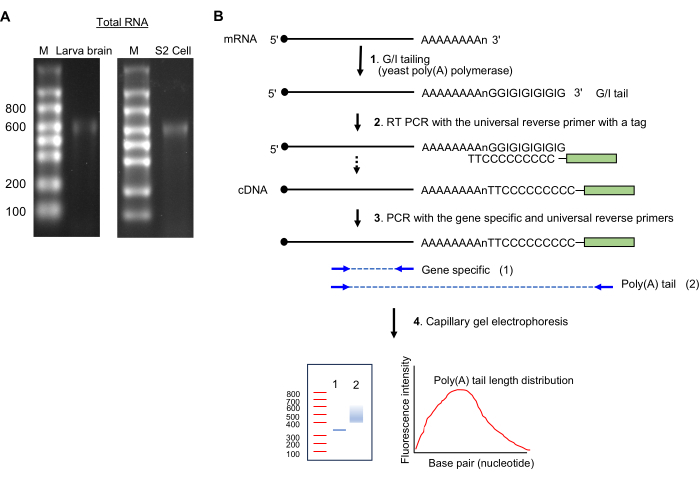
Figure 2: RNA sample preparation and the poly(A)-tail assay. (A) The RNA gel images show total RNA from the Drosophila larva brain (left) and S2 cells (right) on a 1.5% formaldehyde agarose gel. Single-stranded RNA ladder sizes are shown in nucleotides on lane M. Note a major RNA banding at ~600 nt, which is from rRNA. (B) Schematics of poly(A)-tail assay. Abbreviation: G/I = guanosine/inosine. Please click here to view a larger version of this figure.
- GI tailing (Figure 2B)
- Keeping the reagents on ice, prepare the following mixture (20 µL): up to 14 µL of total RNA sample (1 µg), 4 µL of 5x Tail buffer mix, and 2 µL of 10x Tail enzyme mix.
- Incubate at 37 °C for 60 min in a thermocycler.
- Add 1.5 µL of the tail stop solution; keep on ice for 2 min.
NOTE: Proceed to reverse transcription or store the GI-tailed RNA samples at -80 °C until ready to proceed to reverse transcription.
- Reverse transcription and PCR amplification
- Synthesize cDNA by preparing the mix and incubating it under the conditions described in Supplemental File 1.
- Dilute the cDNA samples and perform PCR to amplify DNA as indicated in Supplemental File 1.
7. PCR product analysis by agarose gel electrophoresis
- Analyze a small portion (2-5 µL) of the PCR products from step 6.2.2 on a 2.5% agarose gel by electrophoresis at 100 V for 45 min for quality control.
- Verify the specificity of PCR for gene-specific and tail-specific reactions by sequencing the gel-extracted PCR bands.
8. Capillary electrophoresis
- Perform high-resolution gel electrophoresis on 1 µL of PCR products (0.5-5 ng/µL) from gene-specific and poly(A)-specific PCR using the bioanalyzer with a high-sensitivity DNA kit. Look for well-resolved peaks indicating a successful run.
9. Data analysis: poly(A) tail length measurement (Figure 3)
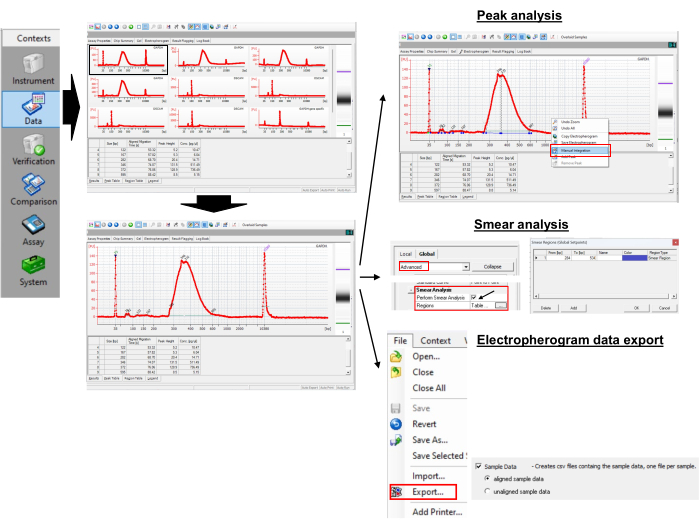
Figure 3: Poly(A) tail length and peak value measurement. Please click here to view a larger version of this figure.
- Accessing data
- To access the data, open the xad file in the software.
- Select the sample name or the ladder in the tree view panel.
NOTE: This will show the result as electropherograms or gel-like images for the selected samples. The lower marker 35 base pair (bp) and upper marker 10,380 bp are the internal standards used to align the ladder data (50-7,000 bp) with data from the sample wells. - Zoom in and out of the electropherograms and gel-like images to display the details.
- Performing peak and smear analysis
- To obtain the peak sizes, open the electropherogram of a selected sample.
- Right-click on the electropherogram and select manual integration to manually select peaks by dragging the horizontal line.
- Observe the peak values in the Peak table. Identify the peak with the largest Peak Height. This is a peak of poly(A) tail length for an individual sample. In the example shown, it is 346 bp.
- Enable the Show/Hide Setpoints icon on the top menu and wait for a new panel to pop up on the right side.
- Select Advanced, scroll down to find Perform Smear Analysis, and select the check box. This will add the Region table to the electropherogram tab.
- Select an electropherogram from a sample and go to the Region table, which shows the From [bp] and To [bp] menu. To set the starting and ending [bp], right-click on the electropherogram and select Region to add Add Region.
- Right-click on any cells in the Region table and select Modify Regions to make a small new window pop up where custom regions can be set.
NOTE: For example, we used a region of 300 bp to 550 bp for GAPDH. The gene-specific GAPDH PCR yielded a peak at 265 bp. The universal primer (Table 1) extends the length of poly(A) PCR by 35 bp via annealing to G/I-tailed RNAs. Thus, the first adenine nucleotide on GAPDH RNA starts at 300 bp (265 + 35). We arbitrarily limited the maximum poly(A) tail length to 250 (300 + 250 = 550). From the region table, the program returns the average size within the region as 387 bp. - Use equation (1) to calculate the poly(A) tail length on the mRNA of interest:
Poly(A) tail length = (A – B – 35) (1)
Where A is the average bp of poly(A)-specific PCR product from electropherogram (i.e., 387 bp for GAPDH), B is the peak bp of gene-specific PCR product from electropherogram (i.e., 265 bp for GAPDH), and "35" is the length of universal reverse primer tag.
NOTE: From the calculation above, the average poly(A) tail length of GAPDH is 387 – 265 – 35= 87 bp.
10. Visualizing poly(A) tail length distribution
- Export data as a csv file under File | Export | Sample Data to retrieve sample data.
NOTE: The exported csv file displays run time rather than bp on the X-axis. - Go to an electropherogram of a sample and check on Show Sizes on the Electropherogram tab to automatically convert the region table on Region Table from bp to run time. In the example, 70.39 s to 86.28 s correspond to 300 bp to 550 bp.
- Open the csv file and select the values from 70.39 s to 86.28 s of run time to generate a graph. To visualize bp sizes to the X-axis on the graph, export the electropherogram with bp sizes as an image file and overlay it on the graph generated in the spreadsheet. This will appropriately match the bp sizes on poly(A) tail distribution.
Here, we analyzed the poly(A) tail length of Dscam1 and GAPDH from Drosophila larval brains (Figure 4). Isolated RNAs were visualized on an agarose gel for quality control. A single RNA band at around 600 nucleotide size indicates intact RNA preparation (Figure 2A). RNAs were subjected to the G/I tailing and high-resolution capillary electrophoresis using an Agilent 2100 bioanalyzer. The gel images were exported using the Agilent 2100 Expert Software and assembled accordingly. The PCR products from GAPDH and Dscam1 gene-specific primer pairs showed a distinct single band while those from the gene-specific/universal primer pairs showed distinct smear patterns (Figure 4A and Table 1), which indicates differential poly(A) length of GAPDH and Dscam1 mRNAs. In Figure 4B, the smear analysis was used to obtain average poly(A) tail lengths from GAPDH and Dscam1. The electropherograms were exported and modified to represent the poly(A) tail lengths (Figure 4C).
Similarly, we performed a poly(A) tail length analysis on S2 cells (Figure 5). To determine the role of Dscam1 3'UTR in shortening poly(A) tails, S2 cells were transfected with the DNA plasmids containing Dscam1 coding region with either SV40 3'UTR or Dscam1 3'UTR. The minimum SV40 3'UTR was selected for control of Dscam1 3'UTR since SV40 3'UTR has been widely used as a minimum transcription termination and polyadenylation signal for recombinant DNA plasmids. Total RNA was extracted and subjected to RT-PCR, G/I tailing, and high-resolution capillary electrophoresis (Figure 5). The result shows that the poly(A) tail length distribution from the SV40 3'UTR plasmid followed a similar pattern as that of endogenous GAPDH while the poly(A) tail length distribution from the Dscam1 3'UTR plasmid followed a similar pattern as that of endogenous Dscam1 mRNA from the larval brains.
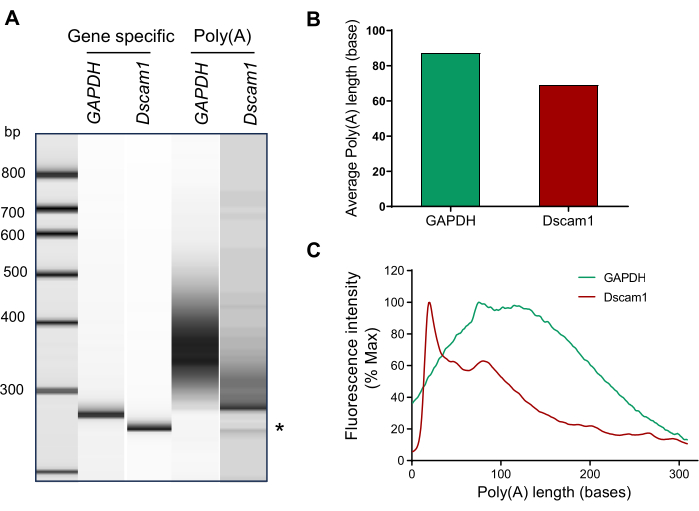
Figure 4: Poly(A) tail length measurement of GAPDH and Dscam1 from the larval brains. (A) Capillary gel electrophoresis resolves the gene-specific PCR product as a discrete band (first and second lanes) for GAPDH2 and Dscam1. The PCR product using universal reverse primer shows a smear distribution (third and fourth lane). * non-specific band. (B) The average poly(A)-tail lengths were calculated using the smear analysis. (C) The distribution of poly(A) tail length was reconstructed from the exported csv file. Please click here to view a larger version of this figure.
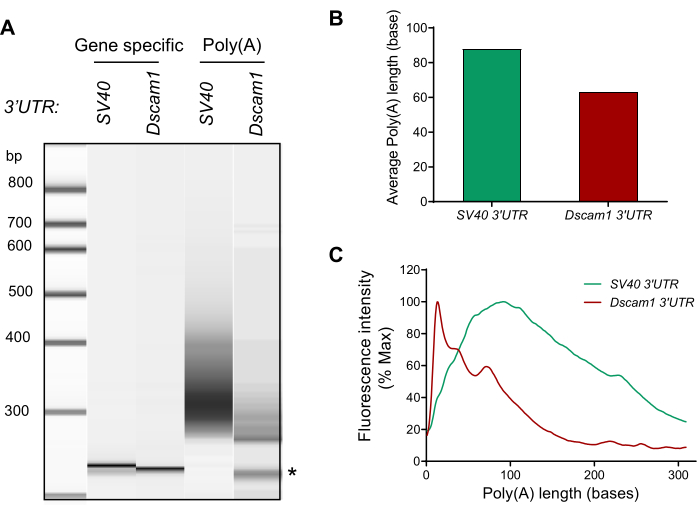
Figure 5: Poly(A) tail length measurement of transfected Dscam1 DNA plasmid. Cultured Drosophila S2 cells were co-transfected with the Dscam1 constructs that contain the coding region of Dscam1, and the 3′UTR of either Dscam1 or SV40. (A) Capillary gel electrophoresis images are shown for the gene-specific PCR product as a discrete band (first and second lanes) for SV40 (Dscam1-SV40 3'UTR) and Dscam1 (Dscam1-Dscam1 3'UTR). The PCR product using universal reverse primer shows a smear pattern (third and fourth lanes). * non-specific band. (B) The average poly(A)-tail lengths were calculated using the smear analysis. (C) The poly(A) tail length distribution was reconstructed from the exported csv file. Please click here to view a larger version of this figure.
| Dscam1 |
| Forward Primer (5’-3’) CGCAGCCACAACAATTGAATG |
| Reverse Primer (5’-3’) AAATAAAATCAAAATCATATATTTAGCAACTTATGAAC |
| SV40 |
| Forward Primer (5’-3’) CCACAAAGGAAAAAGCTGCAC |
| Reverse Primer (5’-3’) TTTATTTGTGAAATTTGTGATGCTATTGCTTTATTTG |
| GAPDH |
| Forward Primer (5’-3’) CACTTCAGAAACGGCCTGAAAATGGC |
| Reverse Primer (5’-3’) AATATTTAAATGCTTATGAGTCGGCATTTTTAAAACTAC |
| Universal reverse primer |
| Reverse Primer (5’-3’) GGTAATACGACTCACTATAGCGAGACCCCCCCCCCTT |
Table 1: Primers used in this protocol.
Supplemental File 1: Composition of solutions and PCR setup. Please click here to download this File.
