The protocols for FLIM sample preparation do not differ from those for confocal or wide-field intensity-based fluorescence microscopy. The data acquisition is followed by the main task of data analysis, i.e. extracting the fluorescence lifetimes from the raw data. Once these have been obtained, data interpretation helps to verify or falsify hypotheses.
1. Staining cells with molecular rotors
- Prepare a stock solution (10 ml) by dissolving approximately 1 mg/ml of the dye in an appropriate solvent (e.g. methanol for BODIPY-C12)6,7 using an accurate balance and a pipette.
- The cells (a model cancer cell line, HeLa in our case) to be stained are grown on in a multiwell plate with a coverslide underside for microscopy, in an incubator at 37 °C with a 5 % CO2 atmosphere until ~ 80% confluent.
- Add 10 – 20 μl of the stock solution to the living cells growing in a multi-well plate (SmartSlide 50 micro-incubation system, Wafergen) in 4 ml of Opti-MEM medium (GIBCO) per well for a 6-well plate. This yields a micro-molar dye concentration in the well.
- Return the multi-well plate to an incubator at 37 °C with a 5 % CO2 atmosphere for 10-45 mins for staining.
- Remove the multiwell plate from the incubator and wash the cells 3-4 times with 4 ml optically clear cell culture medium (e.g. Opti-MEM) to remove excess dye.
- Transfer the multiwell plate to the microscope stage and connect to a temperature controller / 5 % CO2 gas inlet as required, in preparation for imaging.
2. FLIM of fluorescent molecular rotors in cells
- Place the sample on the microscope stage and obtain a transmission and fluorescence image to identify fluorescent cells. A schematic diagram of the experimental set-up is shown in Fig. 1.Verify that the fluorescence emanates from the locations expected (e.g. cell membrane, cytoplasm). Obtain a fluorescence emission spectrum, and verify that it is that of the dye or protein expected, in this case the spectrum of the molecular rotor. As a negative control, image a non-stained sample and verify that it does not fluoresce. Although this step is not essential specifically for FLIM, it is good practice in general and does help to verify that the sample is what you think it is.
- Switch to FLIM mode – this is easily accomplished by moving a mirror out of the fluorescence detection beam path (“external detector” button on “beam path setting” panel on the Leica TCS SP2 acquisition control software). An appropriate fluorescence emission filter to block any exciting light from reaching the detector must be in the fluorescence detection beampath.
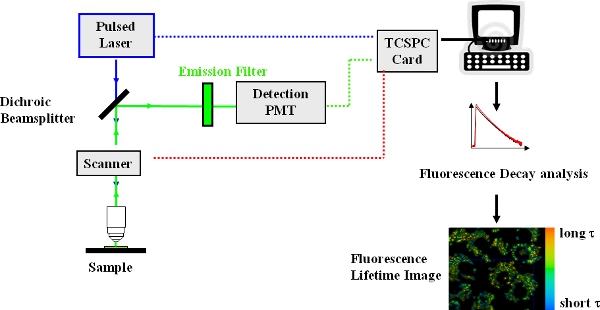
Figure 1. Experimental arrangement for time-domain FLIM using a confocal laser scanning microscope.
- Scan the sample and check, on the computer controlling the FLIM acquisition, that the detector count rate (black bar labeled CFD on acquisition control software of the Becker & Hickl SPC 830 board) is no more than about 1% of the laser repetition rate (green bar labeled SYNC acquisition control software). If it is, reduce the laser excitation intensity, e.g. by placing a neutral density filter in the laser beam path, to avoid collecting pile-up distorted fluorescence decay curves.
- Acquire a FLIM image, typically for 3-5 min, stop scanning and save the raw data (a 3D data “cube” consisting of spatial coordinates x and y, and time).
- Open the raw data in the fluorescence decay analysis software package, for example TRI-214 or commercial software, to display the fluorescence intensity image. This is simply the integrated fluorescence decay, i.e. the area under the fluorescence decay curve, in each pixel.
- Select a typical pixel by placing the cursor on it, and inspect the fluorescence decay in that pixel. If the peak count is below 100, use spatial binning of pixels. The counts of adjacent pixels (e.g. 3×3 or 5×5) are added into the central pixel, so that a higher peak count is obtained there. This provides a higher statistical accuracy for the next step. Alternatively, the measurement could be repeated for a longer acquisition time (step 5). For 30-50min, an approximately 10 times higher peak count (and total counts) is obtained, but this is far too long an acquisition time for most biological samples because of the danger of introducing artifacts due to sample movement, microscope drift, phototoxicity and photobleaching.
- Select a global pixel threshold value (above which the decay in a pixel is fitted) and apply a single exponential decay fit to the image. The result yields a fluorescence lifetime for each pixel above the threshold, which is then encoded in color. Each pixel is colored with the result of the fit, and a FLIM map is obtained. Check the reduced chi-squared values for various pixels – around 1 (and up to 1.3) indicates a good fit. Inspect the corresponding residuals, which should be randomly distributed around zero.
- The fluorescence lifetime histogram plots how often certain fluorescence lifetimes occur versus the fluorescence lifetime itself. Adjust the colour range such that the fluorescence lifetime distribution fits into the colour range.
- If a monoexponential fit does not yield a chi-squared value of around 1 (and up to 1.3), and there is a systematic deviation of the residuals from zero, a more sophisticated model is required. For example, try fitting a double exponential model to the fluorescence decays, to account for two different environments the probe may be in. The fit will also yield the pre-exponential factors or amplitudes which give an indication of the relative amount of dye in one environment or the other. Alternatively, a stretched exponential function may be appropriate to account for a distribution of fluorescence lifetimes.
- The results for the fluorescence lifetimes, pre-exponential factors, and the lifetime ratio and the pre-exponential factor ratio for each pixel can then be encoded in color. Each pixel is coloured according to its value, and contrast due to fluorescence lifetimes, pre-exponential factors and their ratios is obtained. Again, check the reduced chi-squared values (which can also be encoded in colour and displayed as an image) – around 1 (and up to 1.3) indicates a good fit. Inspect the residuals, which should be randomly distributed around zero.
- Fluorescence lifetime histograms should accompany all images for easy visualisation of average fluorescence lifetime values, and the fluorescence lifetime distribution.
3. Representative Results
Fluorescence decays measured for the fluorescent molecular rotor at increasing viscosity in methanol/glycerol mixtures are shown in Fig. 2. The fluorescence decays are monoexponential, and the fluorescence lifetime varies markedly as a function of viscosity. It increases from around 300 ps in methanol (viscosity 0.6 cP) to 3.4 ns in 95% glycerol (viscosity 950 cP).
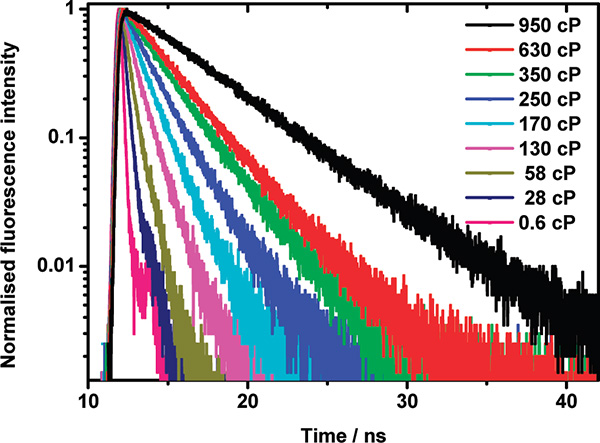
Figure 2. Fluorescence decay profiles for BODIPY-C12 in methanol/glycerol mixtures of varying viscosity.6
The logarithmic calibration plot of fluorescence lifetime τ versus viscosity η for the fluorescent molecular rotor is shown in Fig. 3. It is a straight line as demanded by the Förster Hoffman equation9
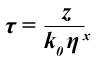
where k0 is the radiative rate constant, and z and x are constants, with 0<x<1. Taking the logarithm on both sides yields
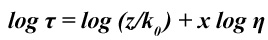
where x is the gradient of the straight line.
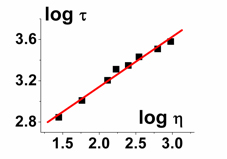
Figure 3. A plot of log fluorescence lifetime vs log viscosity for BODIPY-C12 yields a straight line in accordance with the Förster-Hoffmann equation.6
Following incubation of living cells with the fluorescent molecular rotor a punctate dye distribution is observed in the fluorescence images. FLIM images of HeLa cells incubated with a meso-substituted BODIPY dye are shown in Fig. 4. The fluorescence decays in every pixel of the image can be adequately fitted using a single exponential decay model.
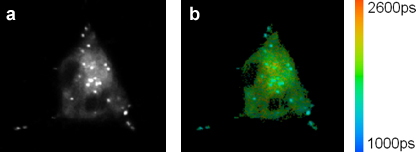
Figure 4. (a) Fluorescence intensity and (b) FLIM images of HeLa cells stained with BODIPY-C12. The bright, punctuate regions exhibit a shorter lifetime than other regions. This shorter liftime corresponds to a lower viscosity in the puncta, probably lipid droplets, according to the Förster-Hoffmann equation.
By plotting the lifetimes extracted from every pixel, we obtain a fluorescence lifetime histogram of the whole image as shown in Fig. 5.
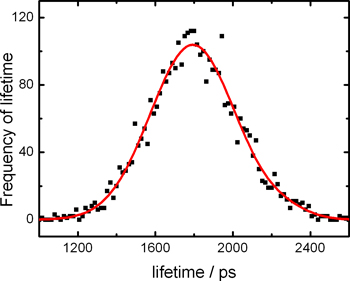
Figure 5. Histograms of fluorescence lifetimes from FLIM images of HeLa cells stained with meso-substituted BODIPY molecular rotors.
