1. Preparation of the RNA library
- The starting DNA library contained 50 nucleotides of random sequences and was synthesized by Integrated DNA Technologies (Coralville, Iowa). The single stranded DNA oligo library sequence is 5′- GGG AGG ACG ATG CGG – N50– CAG ACG ACT CGC CCG A – 3′ (81 nt). The random region is flanked by constant regions, which include the T7 promoter for in vitro transcription and a 3′ tag for RT-PCR. The 5′ and 3′ constant sequences are 5′ – TAA TAC GAC TCA CTA TAG GGA GGA CGA TGC GG – 3′ (32 mer) and 5′ -TCG GGC GAG TCG TCT G – 3′ (16 mer), respectively. Make stock solution with water and store in aliquots at -20°C.
- Amplify the single stranded DNA oligo random library (0.4 μM) by PCR using 3 μM each of 5′- and 3′-primers, along with 2 mM MgCl2 and 200 μM of each dNTP. In order to preserve the abundance of the original DNA library, limit PCR to ten cycles. After the PCR reactions (10 reactions, 100 μL per reaction), recover the amplified dsDNA pool using a QIAquick Gel purification Kit (QIAGEN).
- Convert the resulting dsDNA to an RNA library using the DuraScription Kit (Epicentre, Madison, WI) according to the manufacturer’s instructions. In the transcription reaction mixture, replace CTP and UTP with 2′-F-CTP and 2′-F-UTP to produce ribonuclease resistant RNA. Typically, prepare 20 μL of reaction containing 1 μg of purified DNA template, 2 μL 10 x buffer, 2 μL dATP, 2 μL dGTP, 2 μL 2′-F-dCTP, 2 μL 2′-F-dUTP, 2 μL DTT and 2 μL T7 RNA polymerase at room temperature and then incubate at 37°C for 6 h. (50 mM of each dNTP.)
- Subsequently digest the reaction with DNase I (1.5 μL per 20-μL T7 transcription reaction) to remove the template DNA and purify by an 8% polyacrylamide/7 M urea gel. Quantify the purified RNA library by UV spectrophotometry.
2. In vitro generation of aptamers
- Before selection, prepare the selection buffer and refolding buffer. Prepare a HEPES buffer containing 100 mM Hepes, pH 7.4. Use NaOH to adjust pH value and then store at room temperature. Prepare a RNA refolding buffer (5xHBS) containing 50 mM Hepes pH 7.4, 750 mM NaCl; 5 mM MgCl2; 5 mM CaCl2; 13.5 mM KCl. Prepare a low-salt RNA binding buffer (10 mM HEPES pH 7.4, 50 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2.7 mM KCl, 10 mM DTT, 0.01% BSA and a high-salt RNA binding buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2.7 mM KCl, 10 mM DTT, 0.01% BSA). Store these buffers at -20°C.
- Perform the SELEX principally as described1-4. Before every selection round, refold the RNA pools in 1xHBS buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2.7 mM KCl), heat to 95 °C for 3 min and then slowly cool to 37°C. Continue incubation at 37°C for 10 min.
- Generally, in order to minimize nonspecific binding with the nitrocellulose filters, pre-adsorb the refolded RNA pools to a nitrocellulose filter (HAWP filter, 0.45 μm) for 30 min, prior to incubation with the target HIV-1Bal gp120 protein.
- Incubate the pre-cleared RNA pool with the target protein in low-salt RNA binding buffer for 30 min for SELEX rounds 1 to 4. After the fourth round of SELEX, use a high-salt RNA binding buffer. With the SELEX progress, reduce the amount of gp120 protein and increase the competitor yeast tRNA in order to increase the stringency of aptamer selection.
- For the first cycle of selection, incubate the pre-cleared random RNA pool (40 μg, 1.5 nmol, 9×1014 molecules) and HIV-1Bal gp120 protein (0.23 nmol, RNA/Protein ratio 6.5/1) in 200 μL low-salt RNA binding buffer on a rotating platform at room temperature for 30 min.
- Pass the reaction through a pre-wetted nitrocellulose filter and wash with 1 mL binding buffer.
- Elute the bound RNA from the filter with 200 μL elution buffer (7 M urea and 5 mM EDTA) at 95 °C for 5 min, followed by phenol/chloroform extraction and concentration with a Microcon YM-30 column.
- Reverse transcribe the recovered RNA pool using the ThermoScript RT-PCR system (Invitrogen) and amplify for 15 cycles of PCR.
- Purify the amplified dsDNA pool using a QIAquick Gel purification Kit and transcribe as described above for the next round of selection.
3. SELEX progress monitored by filter binding assay
- Monitor the SELEX progress of the aptamers by filter binding assay. Treat the RNA pool with CIP to remove the initiating 5′-triphosphate and then label with T4 polynucleotide kinase and γ-32P-ATP.
- Heat 10 pmol of CIP treated RNA library at 95°C for 5 min and then chill on the ice. Subsequently, add 2 μL of PNK buffer, 1 μL of T4 polynucleotide kinase, 1 μL of gamma-P32-ATP and water to 20 μL.
- Incubate at 37 °C for 30 min, then add 20 ^mu;L of water and purify the reaction by G-50 column. Finally, obtain 40 μL of labeled RNA at final concentration of 250 nM.
- Before assay, refold the labeled RNA pools in 1xHBS buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2.7 mM KCl), heat to 95 °C for 3 min and then slowly cool to 37°C. Continue incubation at 37°C for 10 min.
- Carry out a 100 μL of binding reaction as example here. Incubate the end-labeled RNA pool (10 nM) with gp120 protein (100 nM) and a 10-fold molar excess of nonspecific competitor tRNA (100 nM) in the high-salt RNA binding buffer for 30 min.
- Separate a 50 μL of binding reaction by a pre-wet nitrocellulose filter.
- Wash the filter with 2 mL binding buffer and count the radioactivity retained on the filter through a multi-purpose scintillation counter (Beckman Coulter). As an input control, count the remaining 50 μL of binding reaction at the same time. Calculate the percent of the RNA retained on the filter in the input RNA as the binding affinity.
4. Cloning, sequencing and alignments
- After 11 rounds, if no further enrichment is observed even following additional selection rounds then maximal binding of the RNA pool has potentially been reached.
- Reverse transcribe the highly enriched aptamer pool (12th RNA pool) using the ThermoScript RT-PCR system (Invitrogen) and subsequently amplify the resulting cDNA by PCR. Purify the PCR product using a QIAquick Gel purification Kit (QIAGEN). Clone the gel-purified the DNA product into the TA cloning vector pCR 2.1 (Invitrogen). In total, inoculate 170 individual clones and further identify them by DNA sequencing to get individual sequences.
- Classify the individual clones into six different groups based on the alignments of individual aptamer sequences. Chose one representative sequence from each group (A-1, A-5, A-9, A-12, A-28 and B-68) for further characterization because of their relative abundance within their group.
5. Generation of aptamer and chimera RNAs by in vitro transcription
- Directly generate double-stranded DNA template by PCR using 2 μM each of 5′- and 3′-primers, along with 2 mM MgCl2 and 200 μM of each dNTP, and recover the resulting PCR products using a QIAquick Gel purification Kit.
- Transcribe chimera sense strand from its PCR generated DNA templates using the DuraScription Kit (Epicentre, Madison, WI). In the transcription reaction mixture, replace the canonical CTP and UTP with 2′-F-CTP and 2′-F-UTP to produce RNA that is resistant to RNase A degradation.
- Typically, incubate 20 μL of reaction containing 1 μg of DNA template, 2 mL 10xbuffer, 2 μL dATP, 2 μL dGTP, 2 μL 2′-F-dCTP, 2 μL 2′-F-dUTP, 2 μL DTT and 2 μL T7 RNA polymerase at 37 °C for 6 h, and subsequently purify it with Bio-Spin 30 Columns (Bio-Rad) following phenol extraction and ethanol precipitation.
- In order to avoid interferon response, further treat the transcribed RNA by CIP to remove the initiating 5′-triphosphate. Incubate total 60 μL of reaction containing 3 μg of transcripts, 6 μL of Buffer 3 and 0.25 μL of CIP at 37 °C for 60 min. After phenol/chloroform exaction and ethanol precipitation, resuspend RNA pellet into water.
- To prepare the chimeras, combine the chimeras harboring only the sense strand RNA with the appropriate antisense RNA in refolding buffer, heat to 95 °C for 3 min and then cool to 37°C slowly. Continue incubation at 37°C for 10 min. Perform refolding step at a finial 1xHBS buffer. For example: mix 10 μL of 10 μM chimera sense strand, 10 μL of 10 μM antisense strand and 5 μL refolding buffer (5xHBS) into 25 μL system.
6. Determination of dissociation constants by gel shift assays
- end P32 label the representative aptamer from each group and chimeras sense strand and then refold RNA in 1xHBS buffer as described above.
- Prepare 25 mL of 5% gel by mixing 2.5 mL of 10xTBE buffer, with 3.125 mL of 40% acrylamide/bis solution, 19.375 mL water, 150 μL of 10% ammonium persulfate (APS) solution, and 30 μL TEMED. The gel should polymerize in about 30 min. Carefully remove the comb and use a 30 -mL syringe fitted with a needle to wash the wells with running buffer (1xTBE).
- Complete the assembly of the gel unit and connect to a power supply. The gel can be pre-run for one hour at 180 V at 4°C.
- Serially dilute the HIV-1Bal gp120 protein with binding buffer to the desired concentrations. The final reaction concentrations of gp120 are 0, 1, 5, 10, 20, 40, 80, 160, 320, 640 nM. Incubate a constant amount of 5′-P32-end-labeled RNA (10 nM) with increased concentrations of gp120 protein in the binding buffer (total 20 μL of reaction) on a rotating platform at room temperature for 30 min.
- After incubation, mix 20 μL of binding reaction with 5 μL native loading buffer and load into a 5% non-denaturing polyacrylamide gel. Prepare a native loading buffer (4x) containing 10 mM Tris-HCl, pH 7.5; 1 mM EDTA, 0.1% Bromophenol Blue, 0.1% Xylene Cyanol FF, 0.1% Orange G, 40% Glycerol. Store in aliquots at -20°C.
- Following electrophoresis (180 V at 4°C for 2 hours, until the secondary dye runs at middle of gel), expose the gel to a Phosphor image screen and quantify the radioactivity using a Typhoon scanner.
- Calculate the dissociation constants using non-linear curve regression with a Graph Pad Prism.
7. Cell-surface binding studies by flow cytometry
- Generate fluorescent aptamer and chimeras using the Silencer siRNA Labeling Kit (Ambion). Add the following reagents in order: 22.5 μL nuclease-free water; 5 mL 10 x Labeling Buffer; 15 μL RNA (5 μg); 7.5 μL Labeling Dye. Incubate total 50 μL labeling reaction at 37°C for 1 hour.
- After incubation, add 5.0 μL (0.1 vol) 5 M NaCl and 125 μL (2.5 vol) cold 100% EtOH, and mix thoroughly. Incubate at -20°C for 60 min. Centrifuge at top speed at 4°C for 20 min. Remove supernatant and wash pellet with 175 μL 70% EtOH. Air dry pellet in the dark and then suspend labeled RNA in 15 μL of nuclease-free water.
- Measure the absorbance of the labeled RNA at 260 nm and at the absorbance maximum for the fluorescent dye. Calculate the base:dye ratio and RNA concentration according to the calculator provided by http://www.ambion.com/techlib/append/base_dye.html.
- Mix Cy3-labeled chimeras sense strand and antisense strand and refold in refolding buffer as described above.
- Obtain CHO-WT expressing gp160 and CHO-EE control cells through the AIDS Research and Reference Reagent Program5, 6. Grow cells in GMEM-S medium (Glutamine-deficient minimal essential medium with 400 μM methionine sulfoximine (MSX)) (Gibco, Invitrogen). Culture the cells in a humidified 5% CO2 incubator at 37 °C.
- Wash the CHO-WT gp160 or CHO-EE control cells with prewarmed washing buffer, trypsinize and detach from the plates. After washing cells twice with 500 μL binding buffer, resupend the cell pellets in binding buffer and incubate at 37°C for 30 min. Pellet cells and then resuspend them in 50 μL of prewarmed binding buffer containing 400 nM Cy3-labeled experimental RNAs.
- After incubation at 37°C for 40 min, wash cells three times with 500 μL of prewarmed binding buffer, and finally resuspend in 350 μL of binding buffer prewarmed to 37°C and immediately analyze by flow cytometry.
8. Internalization and intracellular localization studies by Live-cell confocal microscopy
- Before one day of assay, grow the CHO-WT gp160 and CHO-EE control cells in 35 mm plate with seeding at 0.3×106 in 2 mL GMEM-S medium to allow about 70% confluence in 24 h.
- On the day of the experiments, wash cells with 1 mL of prewarmed PBS. And incubate with 1 mL of pre-warmed complete growth medium for 30 min at 37°C.
- Prepare Cy3-labeled aptamer-siRNA chimeras as described above. Incubate the refold aptamer-stick with the siRNA-stick containing 5′-Cy3-labeled sense strand to form the aptamer-stick-siRNA conjugated as described above.
- Before assay, prepare 0.15 mg/mL solution of Hoechst 33342 (nuclear dye for live cells, Molecular Probes, Invitrogen, CA) in water and store in aliquots at 4°C.
- Stain the cells by treatment with 0.15 mg/mL Hoechst 33342 for 15 min at 37°C. Immediately, wash out dye with 1.0 mL fresh medium twice and replace 2 mL prewarmed fresh medium.
- Add the Cy3-labeled aptamer-siRNA chimera at a 100 nM final concentration into the media and incubate for live-cell confocal microscopy in a 5% CO2 microscopy incubator at 37 °C.
- Collect the images every 15 min using a Zeiss LSM 510 Meta Inverted 2 photon confocal microscope system under water immersion at 40x magnification.
9. In vitro HIV-1 challenge and p24 antigen assay
- Purchase CCRF-CEM cells from ATCC. Grow in RPMI-1640 (Cellgro, Mediatech Inc.) supplemented with 10% fetal bovine serum (FBS, HyClone), L-glutamine and 1xPen-Strep (Gibco, Invitrogen). Culture cells in a humidified 5% CO2 incubator at 37 °C.
- Obtain peripheral blood mononuclear samples from healthy donors from the City of Hope National Medical Center (clinic personnel).
- Isolate PBMCs from whole blood by centrifugation through Ficoll-Hypaque solution (Histopaque-1077, Sigma). Deplete CD8 cells (T-cytotoxic/suppressor cells) from PBMCs by using Dynabeads CD8 (Invitrogen, CA).
- Grow cells in T-cell active medium (BioE, St. Paul, MN). Culture cells in a humidified 5% CO2 incubator at 37 °C.
- Obtain HIV-1 IIIB and NL4-3 virus and HIV-1 Bal virus from the AIDS Research and Reference Reagent Program. After propagation of virus, store in aliquots at -80°C.
- Infect CCRF-CEM cells or human PBMCs with HIV viruses (IIIB, NL4-3 or Bal) (MOI 0.001 or 0.005). After 24 hour of post-infection, gently wash cells with PBS three times to remove free virus. Continue to culture the infected cells in a 5% CO2 microscopy incubator at 37°C for 4 days.
- Prior to RNA treatments, gently wash the infected cells with PBS three times to remove free virus. Incubate 2×104 infected cells and 3×104 uninfected cells with refolded experimental RNAs at 400 nM final concentration in 96-well plates at 37°C (100 μL per well, triplex assay).
- Collect the culture supernatants (10 μL per well) at different times (3 d, 5 d, 7 d and 9 d) and store at -20°C until p24 assay.
- Perform the p24 antigen analyses using a HIV-1 p24 Antigen ELISA kit.
10. The siRNA function detection by quantitative RT-PCR assay
- Infect CCRF-CEM cells or human PBMCs with HIV viruses (IIIB, NL4-3 or Bal) and treat with the experimental RNAs (400 nM) as described above.
- After 7 days of treatment, pellet cells and isolate total RNAs with STAT-60. Treat the total RNAs with DNase I to remove genomic DNA and produce cDNA using 2 μg of total RNA.
- Mix the following reagents: 8 mL nuclease-free water; 1.5 mL 10 x DNase Buffer; 4 μL RNA (2 μg); 0.5 μL RNain inhibitor and 1.0 μL RNase-free DNase I. Incubate total 15 μL reaction 37°C for 1 hour and heat at 80°C for 10 min to inactivate DNase I. Immediately, chill the reaction on the ice.
- Add 2 μL random primer (50 ng/μL) and 1μL dNTP (10 mM) into the reaction mixture above, and then heat at 65°C for 5 min. Immediately, chill the reaction on the ice.
- Add the following reagents: 5 mL 5xFirst strand buffer; 2.5 mL 0.1 M DTT; 0.5 mL RNain inhibitor and 1.0 mL MMLV-RT. Incubate total 27 mL reaction at 25°C for 10 min and at 37°C for 1 hour. After reaction, heat mixture at 70°C for 15 min to inactivate reverse transcriptase and then chill on the ice. The cDNA is ready for qRT-PCR analysis.
- Analyze expression of the target genes by quantitative RT-PCR using 2 x iQ SyberGreen Mastermix and specific primer sets at a final concentration of 400 nM (triplex assay). Use the GAPDH expression as an internal control for normalization of the qPCR data.
11. Representative results:
1. New RNA aptamers against HIV-1BaL gp120 are isolated and characterized.
As described in the experimental section, an initial DNA oligonucleotide library containing a 50 nt random region flanked by fixed primer regions on the 5′ and 3′ ends is amplified and transcribed into an RNA pool. This initial library consists of up to 1015 diverse sequences (1 nmol), which fold into a vast array of various 3-D structures. The high complexity and diversity of the initial library might guarantee the presence of active structures with good binding affinity to the target.
Employ an in vitro SELEX procedure (Figure 1) to select 2′-fluoropyrimidine modified RNA aptamers which selectively bind the R5 strain HIV-1BaL gp120 envelope protein7. As shown in Figure 1, a nitrocellulose-based selection strategy is performed to isolate specific target-binding RNAs from non-binding RNA molecules. Since the protein sticks to nitrocellulose, only the RNA/protein complexes or aggregates can be retained on the membrane and free RNAs are washed out. Under denaturing conditions, the bound RNAs are recovered and are reverse transcribed to cDNA and then amplified into dsDNA, and subsequently in vitro transcribed to create a new RNA pool for next selection cycle. The selection stringency is increased by reducing the amount of target protein and increasing the amount of competitor tRNA. The amount of RNA pool, protein and competitor tRNA used in each selection round is shown in Table 1.
Monitor the progress of selection after each SELEX cycle by the filter binding assay. Evaluate the binding affinity as the percent of the RNA retained on the filter in the total RNA pool. The starting RNA pool (1-RNA) only shows 0.1% of the input RNAs retained on the membrane. However, after nine selection rounds the ninth RNA library (9-RNA) has 9.72% of the input RNA bound. Although additional selection rounds were conducted, no further enrichment is observed, suggesting that maximal binding of the RNA pool has been reached (Figure 2A). Similar with the filter binding assay, the gel shift assay also is one of the most popular strategies for determining dissociation constants. This procedure is easy and convenient. As shown in Figure 2B, gel shift assays further confirm the binding activities of the RNA pools. These results demonstrate that some ligands with high binding specificity for the target protein are successively enriched in these RNA pools..
Clone and sequence the highly enriched aptamer pools (12-RNA). According to the alignments of individual cloned aptamer sequences, six different groups are classified as shown in Table 2. About 40% of the clones (Group I and II aptamers) contain a conserved sequence: A(A/G)TTGAGGGACC(A/G). We choose one representative sequence from each group (for example: A-1, A-5, A-9, A-12, A-28 and B-68) for further characterization because of their relative abundance within their group. Through a native gel mobility shift assay, the dissociation constants (Kd) of these representative aptamers are calculated (Figure 3A). For example, A-1, the best of the aptamers, has an apparent Kd values of 52 nM (Figure 3B). As shown in Figure 3A, these selected aptamers can selectively bind with the target HIV-1 Bal gp120, but not the HIV gp120 CM protein.
2. Anti-gp120 aptamer specifically binds and is internalized by cells expressing HIV gp160 and inhibit HIV-1 infection in cell culture.
CHO-gp160 cells stably expressing the HIV envelope glycoprotein gp160 are used to test for binding and internalization of the selected anti-gp120 aptamers. These cells do not process gp160 into gp120 and gp41 since they lack the gag encoded proteases required for envelope processing. As a control we use the parental CHO-EE cell line which does not express gp160. Flow cytometric analyses (Figure 4A) reveal that the Cy3-labeled aptamers specifically bind to the CHO-gp160 cells but not the control CHO-EE cells. Furthermore, real-time live-cell Z-axis confocal microscopy indicates that the Cy3-labeled aptamer is selectively internalized within the CHO-gp160 cells (Figure 4B and 4C) after 2 hours of incubation, but not the CHO-EE control cells (Figure 4D). Figure 4C also shows that the aptamer aggregated within the cytoplasm, which suggests the gp120 aptamers maybe enter cells via receptor-mediated endocytosis.
In the HIV-1 challenge assay, HIV-1 infected-PBMC cells are treated with the aptamers. At different days post treatment with the aptamers, aliquots of the media are assayed for viral p24 antigen levels (Figure 4E). The results show that the anti-gp120 aptamers (A-1) inhibits HIV-1 p24 production with a nano-Molar concentration.
3. Anti-gp120 aptamer-siRNA chimera is designed and evaluated its efficacy as cell-type specific siRNA delivery system
As shown in Figure 5A, the aptamer and sense strand segment of the siRNAs contained nuclease-resistant 2′-Fluoro UTP and 2′-Fluoro CTP and are synthesized from corresponding dsDNA templates by in vitro bacteriophage transcription. In order to increase the flexibility of the molecule, a two nucleotide linker (UU) is inserted between the aptamer and the Dicer substrate portion. To prepare the siRNA containing chimeras, in vitro transcribed chimeric aptamer-sense strand polymers are annealed with equimolar concentrations of an unmodified antisense strand RNA. These data from gel shift assay (Figure 5B) and flow cytometry (Figure 5C) indicate that the chimeras maintain approximately the same binding affinities as the aptamers alone. The time-course images from real-time confocal microscopy (Figure 5D) show that Cy3-labeled chimera Ch A-1 can be successfully internalized into the cytoplasm of cells. As expected, no uptake of the chimera is observed with the CHO-EE control cells (Figure 5E).
Similarly, the antiviral potential of RNAs is evaluated by HIV-1 challenge assay. The results of HIV p24 antigen analyses (Figure 6A and 6B) show that both aptamer and chimera inhibit p24 production, but the strongest inhibition is observed with the chimera Ch A-1 treatment.
To confirm that the siRNA component is functioning along with the aptamer, following internalization of the Ch A-1 chimera in infected cells, we also evaluate the relative levels of inhibition of tat/rev gene expression by quantitative RT-PCR expression assays. We find that the treatment of infected cells with the chimeras is able to induce silencing of the tat/rev gene, while the aptamer alone did not affect tat/rev gene expression (Figure 6C and 6D). These results provide further support that the aptamer delivered siRNA triggers RNAi.
| The amount of protein, RNA pool and tRNA used for selection | |||||
| SELEX rounds | Ratio of Target/RNA | Gp120 protein | RNA pool | Competitor tRNA | Selection Buffer |
| 1 | 1/6.5 | 229.8 pmol | 1.5 nmol (40.1 μg) | 0 | Low salt SELEX buffer |
| 2 | 1/6.5 | 114.9 pmol | 0.75 nmol (20.1 μg) | 0.25 nmol (6.6 μg) | |
| 3 | 1/8 | 76.6 pmol | 0.625 nmol (16.7 μg) | 0.25 nmol (6.6 μg) | |
| 4 | 1/8 | 76.6 pmol | 0.625 nmol (16.7 μg) | 0.25 nmol (6.6 μg) | |
| 5 | 1/8 | 38.3 pmol | 0.306 nmol (8.18 μg) | 0.5 nmol (13.2 μg) | High salt SELEX buffer |
| 6 | 1/8 | 38.3 pmol | 0.306 nmol (8.18 μg) | 0.5 nmol (13.2 μg) | |
| 7 | 1/10 | 26.8 pmol | 0.268 nmol (7.16 μg) | 0.5 nmol (13.2 μg) | |
| 8 | 1/10 | 26.8 pmol | pmol 0.268 nmol (7.16 μg) | 1 nmol (26.4 μg) | |
| 9 | 1/10 | 15.3 pmol | 0.153 nmol (4.09 μg) | 1 nmol (26.4 μg) | |
| 10 | 1/10 | 15.3 pmol | 0.153 nmol (4.09 μg) | 1.5 nmol (39.6 μg) | |
| 11 | 1/12.5 | 7.66 pmol | 0.096 nmol (2.56 μg) | 2 nmol (52.8 μg) | |
| 12 | 1/12.5 | 7.66 pmol | 0.096 nmol (2.56 μg) | 2 nmol (52.8 μg) | |
Table 1. The selection conditions. The amount of protein, RNA pool and tRNA used for each selection and selection buffer are indicated.
| Group | RNA | Random sequences | Frequency (140 clones) |
| Group I | A-1 | AATTGAGGGACCACGCGCTGCTTGTTGTGATAAGCAGTTTGTCGGATGG | 33 (23.6%) |
| B-7 | AATTGAGGGACCAACGCGAGGATGTGGATAGTGTGTATTTGCGTGATGG | 3 | |
| A-32 | AATTGAGGGACCGTTGGTAAAAGCCGGAAATTGAGCTTTTACGGCGATGG | 5 | |
| B-55 | AATTGAGGTACCGCGTTATTAGGAACAAATTGGAATTCTAAACGCGATGG | 2 | |
| A-24 | AATAGAGGGACCCAGATATAGGCTACACGGATGATGGTGTATCTGGATGG | 1 | |
| B-19 | AATAGAGGAACCGTTTCAGAAGACTACAGGTTAGTCCAATGAAGCGACGG | 1 | |
| B-31 | AATAGAGGGACCGTGGACAATAATTTATGGTCA TTTATTGGCACGATGG | 1 | |
| Group II | A-12 | AGTAGAGGAACCAAGCAATGGATGAATGCAAAAGTGTAAATGCTTGATGG | 10 (7.1%) |
| Group III | A-9 | TGAGTTTGGGTAAATTTCCGGTTTCGGTTTACTCACGAAAGATCGGTCGG | 15 (10.7%) |
| Group IV | A-28 | TAAAGGAGGGAAGGATGAGACCGCACGAAAAATATCAGCATACGTTTGTG | 10 (7.1%) |
| Group V | A-5 | GAAACTAGTTTGAATAATGGTGTAGAGGAGGGTCAATAGTTTCGTTGGTG | 9 (6.4%) |
| Group VI | B-68 | ACATAGTAATGACACGGAGGATGGAGAAAAAACAGCCATCTCTTGACGGT | 2 |
| Others | Orphan sequence | 48 |
Table 2. The alignment and identification of RNA aptamers. Following the 12th round of selection, the selected RNA pool was cloned and sequenced. After alignment of all 140 clones, six groups of anti-gp120 aptamers were identified. Only the random sequences of the aptamer core regions (5′-3′) are indicated. Isolates occurring with multiple frequencies are specified.
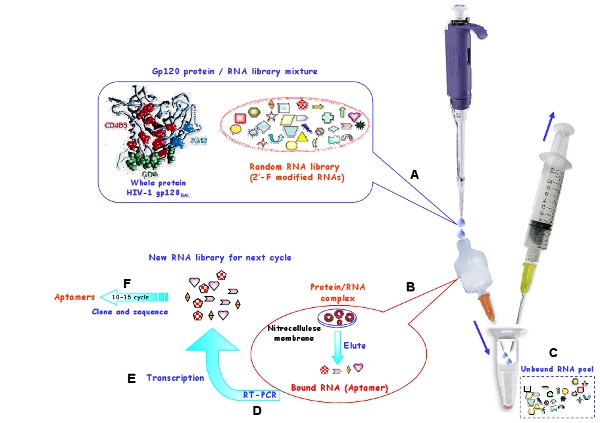
Figure 1: Schematic representation of the in vitro selection procedure using a nitrocellulose membrane, for generating RNA aptamers for HIV-1 Bal gp120 protein. (A) The starting RNA pool and target protein were incubated to form complex. (B) The bound RNA molecules were retained on the membrane and eluted from the membrane under denaturing condition. (C) The unbound RNAs were washed away. (D) The selected RNAs were reverse transcribed and amplified by PCR. (E) The relevant DNA was subsequently transcribed into new RNA pool for next selection cycles. (F) After 10-15 selection rounds, the selected aptamers were cloned and sequence.
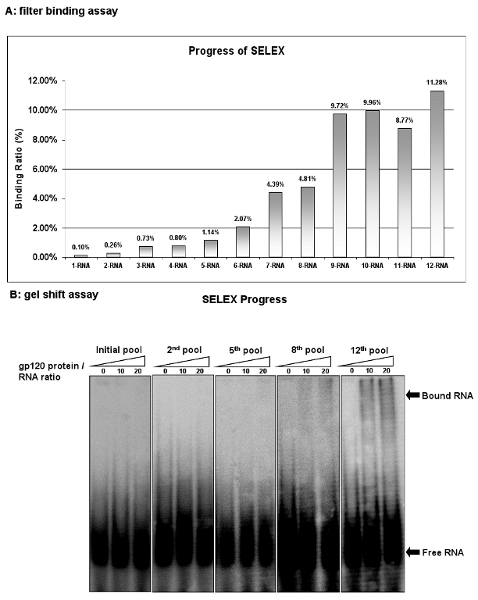
Figure 2: The progress of HIV-1 gp120 aptamers selection. (A) The binding activity of the RNA pool at each cycle was analyzed by filter binding assay with competitor tRNA. Binding activities were calculated as the percentage of input RNA retained on the filter in protein/RNA complex. (B) The binding activity of the RNA pool at each cycle was analyzed by gel shift assay. The 12th RNA pool showed the highest binding activity.
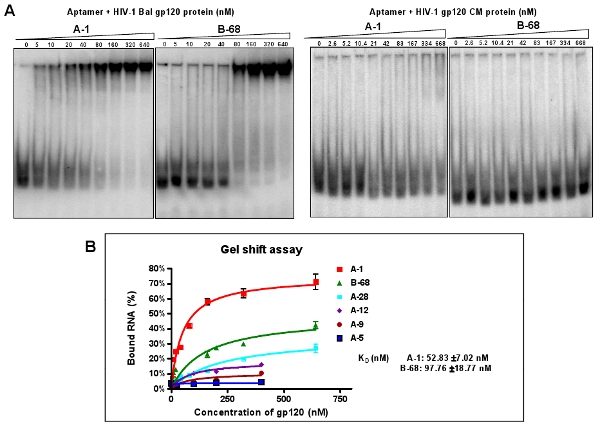
Figure 3: Binding activity assay of selected individual aptamers against HIV-1Bal gp120. (A) The 5′-end P32 labeled individual aptamers were incubated with the increasing amounts of target gp120 protein or non-specific CM protein. The binding reaction mixtures were analyzed by a gel mobility shift assay. Aptamer A-1 and B-68 showed the best binding affinity with the target protein, but not CM protein. Data represent the average of four replicates. (B) Binding curve from a gel shift assay.
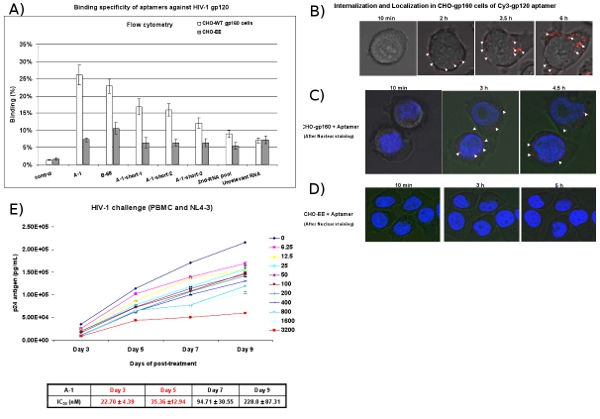
Figure 4: Cell-type specific binding and uptake studies of aptamers. (A) Cell surface binding of Cy3-labeled RNAs was assessed by flow cytometry. Cy3-labeled RNAs were tested for binding to CHO-gp160 cells and CHO-EE control cells. The selected aptamers showed cell-type specific binding affinity. The 2nd RNA pool and irrelevant RNAs were used as negative controls. Data represent the average of three replicates. (B) Internalization analysis. CHO-gp160 cells were grown in 35 mm plates and incubated with a 100 nM concentration of Cy3-labeled A-1 in culture media for real-time live-cell-confocal microscopy analysis. The images were collected at 15 min. intervals using 40X magnification. (C, D) Localization analysis. CHO-gp160 cells and CHO-EE control cells were grown in 35 mm plates. Before incubation with 100 nM of Cy3-labeled A-1, cells were stained with Hoechst 33342 (nuclear dye for live cells) and then analyzed using real-time confocal microscopy. (E) The selected anti-gp120 aptamers inhibit HIV-1 replication in human PBMCs previously infected with HIV-1 NL4-3 virus. Different concentrations and time pointes were presented. IC50 value was listed. Data represents the average of triplicate measurements of p24.
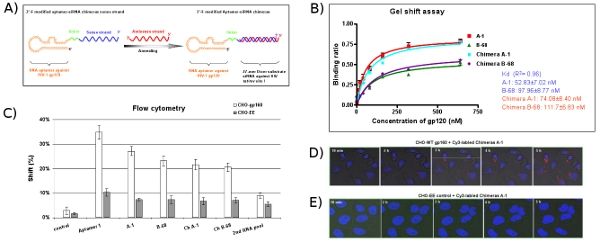
Figure 5: The design and evaluation of the aptamer-siRNA chimera delivery system. (A) Schematic aptamer-siRNA chimeric RNAs: the region of the anti-gp120 aptamer is responsible for binding to gp120 and the siRNA is targeting a common exon of HIV-1 tat/rev. The 2′-Fluoro modified aptamer-siRNA sense single strand was co-transcribed, followed by annealing of the complementary siRNA antisense strand to complete the chimeric molecule. A linker (UU) between the aptamer and siRNA is indicated in green. (B) The aptamer-siRNA chimeric RNAs that have comparable Kd values as well as parental aptamers specifically bind the HIV Bal gp120 protein. Data represent the average of three replicates. (C) Cell-type specific binding studies of aptamers. Cy3-labeled RNAs were tested for binding to CHO-gp160 cells and CHO-EE control cells. Cell surface bindings of Cy3-labeled RNAs were assessed by flow cytometry. The selected aptamers showed cell-type specific binding affinity. The 2nd RNA pool and irrelevant RNA were used as negative controls. Data represent the average of two replicates. (D, E) Internalization and intracellular localization analyses. CHO-gp160 cells were grown in 35 mm plates and were stained with Hoechst 33342 (nuclear dye for live cells). Subsequently, cells were incubated in culture medium with a 100 nM concentration of Cy3-labeled chimera for real-time live-cell confocal microscopy analysis as previously described.
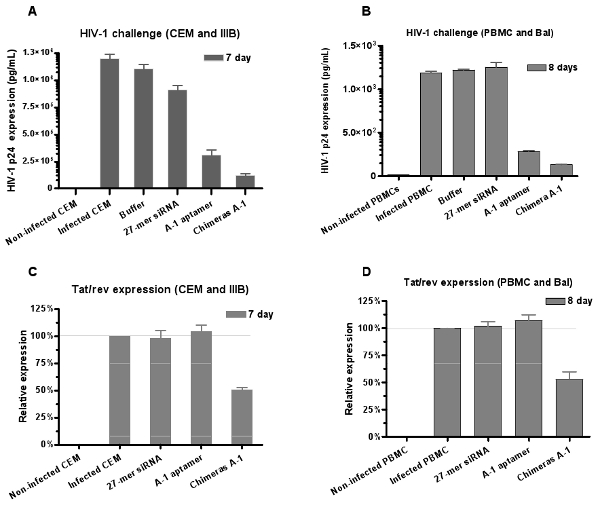
Figure 6: Dual inhibition of HIV-1 infection mediated by aptamer-siRNA chimeras. Both anti-gp120 aptamer and aptamer-siRNA chimeras neutralized HIV-1 infection in (A) CEM cells (IIIB strain) and (B) human PBMCs (BaL strain) culture, respectively. Data represent the average of triplicate measurements of p24. The chimeras showed stronger inhibition than aptamer alone indicating that (C, D) the siRNA delivered by the aptamers down-regulated tat/rev gene expression in the PBMCs. Data represent the average of three replicates.
