1. Microchip fabrication
- SU8 master fabrication.
To obtain microfluidic channels of 40 μm height, spin coat SU8-50 (MicroChem Corp.) on a Si wafer at 3000 rpm for 30 s. Soft bake on a hot-plate at 65°C for 5 minutes and 95°C for 30 minutes to evaporate the solvent and densify the film. After cooling down, expose the SU8 resist with 360-440 nm radiation at 300 mJ/2 under the appropriate mask. Following exposure, bake the wafer at 65°C for 2 minutes and 95°C for 4 minutes. After cooling, develop the unexposed areas (Micrpoposit EC solvent, Chestech) for 5 minutes, with stirring.5 Use fresh EC solvent to rinse the wafer and then dry it under gas to generate a clean surface. Treat the wafer with hexane and methanol washes. Complete the SU8 master fabrication process with a final step of evaporating trichloro(1,1,2,2-perfluoocytl)silane onto the surface (in a desiccator for 40 minutes). - PDMS curing, dicing and hole-punching.
To form structured microfluidic substrates, we mix Sylgard 184 (Dow Corning) in a 10:1 (w/w) ratio of resin to crosslinker and then pour over the master. Degas in a desiccator, and then cure the device (top layer) for one hour at 65°C. Cut the cured PDMS and peel it off the master. Create access holes for fluidic tubing using a biopsy punch. Cure another layer of PDMS (8 g of the resin evenly spread in a 10×10 cm Petri dish) for 20 minutes at 65°C. Place the prepared top layer on the bottom layer and then cure overnight at 65°C. Peel the chips from the Petri dish and dice for use.
2. Experimental setup
- Microfluidic setup
- Fill syringes (plastic or glass from Beckton, Dickinson and Company or from SGE Analytical Science) with the required solutions, ensuring no air bubbles remain in any parts of the tubing. For droplet generation two phases are used. The aqueous (dispersed) phase contains the reagents of interest (for example a suspension of cells in growth medium, or a solution of molecular fluorophores). The oil phase, which in this case acts as the continuous phase, is generally formed from a mixture of oil and surfactant, e.g. 2% Raindance surfactant (RainDance Technologies) in fluorinated oil (3M Fluorinert Electronic Liquid FC-40); or 2% Span 80 (Croda) in Mineral oil.
- Fit tubing (Polyethylene tubing, Harvard Apparatus) of the same internal diameter as the syringe needle tips on one end to the needles. The tubing should be cut to the shortest possible length to minimize flow instabilities due to vibrational perturbations.
- Connect the other ends of the tubing directly to the holes punched in the PDMS substrate. More complex solutions, such as the use of silica capillary ports or connectors (e.g Upchurch Nanoports) can also be implemented for a more robust interface between the microfluidic device and the tubing. Connect the outlet port of the chip to tubing and direct into a collector (for waste collection or further analyte processing).
- Mount the syringes on a syringe pump (e.g. PHD 4400 Hpsi programmable syringe pumps, Harvard Apparatus) and define the desired infusing flow-rate for each phase (usually on the order of microliters per minute). A minimum ratio of oil/aqueous flow-rates of 1 is recommended to avoid wetting of the PDMS by the aqueous phase, which would lead to unstable droplet formation (depending on device geometry). For the same reason, it is also recommended to first flush the oil phase alone, through any microfluidic channels, before introduction of the aqueous phase. The final setup used for the experiments herein is illustrated in Figure 2a.
- Optical system setup
- To probe small volumes (down to a few picoliters) within the microfluidic channels with high spatial and temporal resolution use a confocal microscope (with automated submicron positioning capacity).
- Use water-soluble fluorescent molecules with ideally high quantum yields. Excite the molecules using a laser operating at a wavelength that matches the absorption of the fluorophore involved. For example, common fluorescent molecules such as fluorescein 5-isothiocyanate (FITC) and Alexa-Fluor 488 can be efficiently excited using a 488 nm diode laser (e.g. PicoQuant Gmbh, LDH-P-C-485). Fluorescent molecules, such as Texas Red or Allophycocyanin (APC) can be efficiently exited at 633 nm using a He/Ne laser (e.g. Newport R-31425).
- Use pulsed lasers operating at repetition rates of several MHz (with several ns pulse separations) and fluorophores with sufficiently differentiated lifetime properties for Fluorescence Lifetime Imaging (FLIM) experiments.
- Use beam-steering mirrors and optics to control the beam height as well as beam direction.
- Use a dichroic mirror to reflect the laser beam into the objective lens. If two lasers are used simultaneously, a dual-band dichroic mirror can be installed to allow reflection of the two laser beams (e.g. for 488 and 633 nm beams a z488/633rdc mirror from Chroma Technology Corporation is ideal).
- Use an infinity-corrected, high numerical aperture (NA) microscope objective (i.e. Olympus 60 x /1.2 NA, water immersion) to bring the laser light to a tight focus within a microfluidic channel. When working with tall microchannels (>100 μm) the objective used must be appropriately selected to ensure that its working distance effectively covers the full height of the microchannel.
- Collect fluorescence emitted by the sample (within the microfluidic channel) using the same high NA objective and transmitted through the same dichroic mirror.
- Remove any residual excitation light using a single or dual-band emission filter (e.g. a z488/633 filter from Chroma Technology Corporation).
- Focus the fluorescence emission onto a 75 μm pinhole using a plano-convex lens (e.g. +50.2 F; Newport Ltd.). Position the pinhole in the confocal plane of the microscope objective.
- Use another dichroic mirror (e.g. 630dcxr, Chroma Technology Corporation) to split the fluorescent signal onto two detectors.
- Filter the fluorescence reflected by the dichroic mirror with an emission filter (e.g. an hq540/80 m filter from Chroma Technology Corporation) and focus it with a plano-convex lens (e.g. f = 30.0, i.d. 25.4 mm, Thorlabs) onto the first (‘green’) detector. For experiments involving a secondary red fluorophore filter the fluorescence transmitted by the dichroic mirror with another emission filter (e.g. an hq640lp filter from Chroma Technology Corporation) and then focus it with another plano-convex lens onto the second (‘red’) detector.
- Use avalanche photodiodes (e.g. AQR-141, EG&G, Perkin-Elmer) for detection of fluorescence photons. The final setup is illustrated in Figure 2b.
3. Droplet generation and data acquisition
- Droplet generation methods
- You can use two main configurations of microchannels to generate droplets: a flow focusing or T-junction format (Figure 3a and 3b).6,7 Both methods are equivalent as long as the droplets formed are as wide as the microchannel itself. As a result of the shear forces that arise from using these geometries to bring the two immiscible fluids together and the subsequent capillary instabilities that develop in the interface, monodisperse droplets (as small as a few femtoliters) are generated spontaneously.
- You can encapsulate reagents in different ways: reagents can be prepared and mixed off chip to the desired mixture/concentration, or they can be brought separately into the chip and then combined together prior to droplet formation (Figure 3c). This last method provides for higher flexibility since mixtures/concentrations can be modified by simply tuning the relative flow-rates. It is noted that for cell encapsulation, the cell suspension in the syringe should be stirred to avoid condensation of the cells over the timescale of the experiment.
- Fluorescence data acquisition.
- Couple the electronic signal from the avalanche photodiode detector (described in 2.2.12) to a multifunction DAQ device for data logging (e.g. a PCI 6602, National Instruments), running on a personal computer.
- For FLIM experiments connect the detectors to a Time-Correlated Single Photon Counting (TCSPC) card (e.g. TimeHarp 100, PicoQuant GmbH) running on a separate PC. This card allows for fluorescence lifetime data to be obtained using a TCSPC methodology: each detected fluorescent photon is correlated to an incident laser pulse, and therefore each emitted fluorescent photon is assigned a delay time. This data can be fitted to a single or multi-exponential decay curve to obtain an estimation of the fluorophore/s radiative rate coefficient/s. Deconvolute the raw data with the instrument response function (IRF) of the detector: this is obtained using a low concentration Auramine-O solution (which exhibits a fluorescence lifetime of a few picoseconds).8
4. Cell recognition and mapping of mixing within droplets
- Cell recognition and mapping of mixing within droplets
- Use Escherichia coli Top 10 strain for the viability assay experiment. Culture the cells in Luria-Bertani broth overnight and match the optical density to 0.5 prior to the experiments.
- Use 0.4 μM SYTO9 and 1 μM propidium iodide for detecting the viability of the cells. Both are DNA-intercalating dyes and their fluorescence intensity increases by over 20 folds upon binding to DNA. SYTO9 is a green fluorescent dye that is membrane permeable and propidium iodide is a red fluorescent dye which is membrane impermeable. Thus live cells fluoresce ‘green’ while dead cells exhibit both ‘green’ and ‘red’ emissions.
- Use the setup introduced in 2.2) for green/red fluorescence detection.
- Use a portable, mini-magnetic stirrer (Utah Biodiesel Supply, Utah) to stir the cell suspensions within a 3mL BD plastipak syringe fitted with a 7mm magnetic stir bar to prevent cell sedimentation .
- Mapping of mixing events using FLIM
- Focus the optical probe volume at half the height of a microchannel along which droplets are flowing.
- Form droplets from two aqueous solutions (as in Figure 3c), each containing a (non-interacting) fluorophore with different characteristic fluorescent lifetimes.
- Beginning from one side of the channel, carry out each experiment along the entire width of the channel at 1 μm intervals. The channel edges can be easily identified as the fluorescence intensity drops drastically once the laser beam is focused in their proximity.
- Implement an algorithm to differentiate signal bursts (associated with droplets) from the noise background of the oil phase and to establish the duration of each burst.
- Implement a second algorithm to extract the delay time and intensity values along the length of each droplet at the particular width where the experiment has been carried out.9
- Then use a Maximum Likelihood Estimator (MLE) algorithm to evaluate the fluorescence lifetime for each droplet in the experiment.8 Averaging the lifetime values for all the droplets in the experiment, reduces the final error of the MLE calculation (the more droplets probed, the smaller the error).
- Once a lifetime trajectory has been obtained for each width, combine all the trajectories in a 2D map. Since each lifetime value is associated with a particular mixture of the two fluorophores, a concentration (or mixing) map can thus be obtained.
- Optionally, a 3D map of droplet mixing could be easily obtained by repeating this protocol at different positions along the height of the microfluidic channel.
5. Data analysis
- Reinterpret the raw experimental data files into the appropriate computing language so that they can be further analyzed (e.g. text files that contain the variation of photon counts over time, can be readily extracted into Matlab for data processing and analysis). You might have to write specific codes in order to access the data contained in files of more complex experiments (such as those for FLIM) or in order to run MLE algorithms or build 2D maps of mixing patters from lifetime trajectories.
6. Representative Results
Cell recognition
Typical examples of the variation of photon counts as a function of time for the cell-based experiments are shown in Figures 4a and c, over a period of two hundred milliseconds. Importantly the Luria–Bertani broth (LB medium) acts as a weakly fluorescing background defining the aqueous droplet boundaries, whilst distinct photon bursts, corresponding to the presence of individual cells, can be distinguished on top of this background. The LB fluorescent background is characterised by an approximately rectangular cross-section (marked by the red and green dotted lines).
The full width half maximum (FWHM) of fluorescent bursts arising from E. coli cells is 30-fold shorter than the background droplet events, which is consistent with the relative length of droplets (40 microns, corresponding to a volume of 30 picolitres) and E. coli cells (1.5–2.0 microns).10
With the dual channel detection system, the existence of live cells or dead cells can be distinguished from analysis of the green and red channels. Figures 4 b and d show the probability distribution describing the number of cells per droplet.
Mapping of mixing events using FLIM
The molecular fluorescence lifetime is an intrinsic property of an individual fluorophore that is affected only by its chemical environment. Accordingly its measurements can be used to obtain environmental information (such as solution pH, temperature, polarity and viscosity) with superior resolution and precision than time integrated fluorescence measurements, which are dependent on experimental factors (such as concentration, excitation intensity, and optical collection efficiency).9,11
As presented elsewhere,8 it is possible to map the mixing patterns inside droplets by using two fluorophores with different lifetime values. In a mixture containing two (non-interacting) fluorescent species the decay will take on a biexponential form as is shown in equation 1:
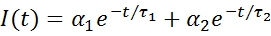
The individual lifetimes are defined as τ1 and τ2; and the amplitude of each component is given by α1 and α2 respectively. The average lifetime can then be defined as follows:
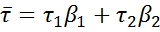
Where β1 and β2 are the fraction of each component and are defined as follows:

Since the probe volume is fixed and droplets are flowing across that position, a trajectory of the lifetime values along the length of the droplets at that particular width can be thus obtained. A characteristic trajectory for a particular width, averaged among 6000 droplets, can be observed in Figure 5a.
Combining the trajectories throughout the entire width of the channel leads to the formation of a 2D concentration (or mixing) map. A typical result is presented in Figure 5b.
By averaging the results among a large number of droplets, the standard error of the results obtained can be greatly reduced (1% of standard error with a 95% confidence interval for Figure 5a). The determination method of the lifetime trajectory assumes that all droplets are practically identical. This is proven to be largely correct for two main reasons: if droplets were significantly different, the averaged lifetime trajectories obtained would have flatter, less diverging profiles (and sharp differences in lifetime along the length of the averaged droplets are consistently detected). Furthermore, the results are reproducible regardless of the individual droplets analyzed or the amount of droplets used in the calculations.8
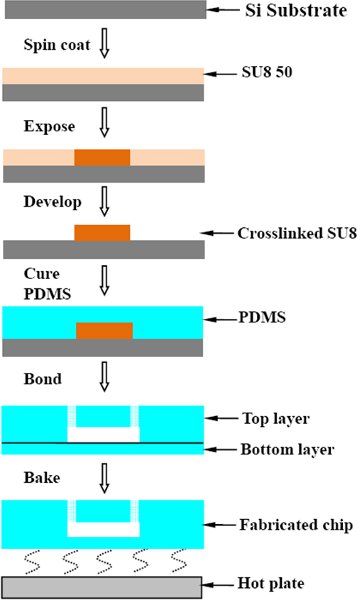
Figure 1. process flow on the microfluidic chip fabrication.
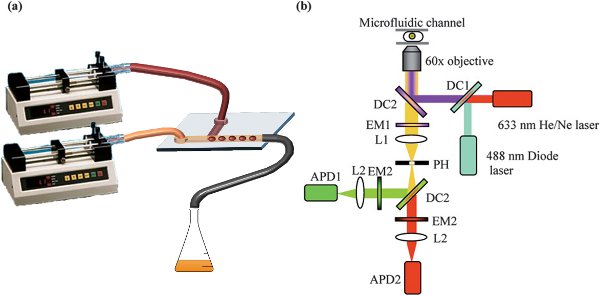
Figure 2. a) Diagram representing the syringe pumps and syringes, connected to the microfluidic chip for droplet formation; b) Schematic representation of a typical optical setup for a two laser – two fluorophore (green and red) excitation and detection. DC = Dichroic mirror, EM = Emission filter, L = Lens, PH = Pin hole, APD = Avalanche photodiode detector.
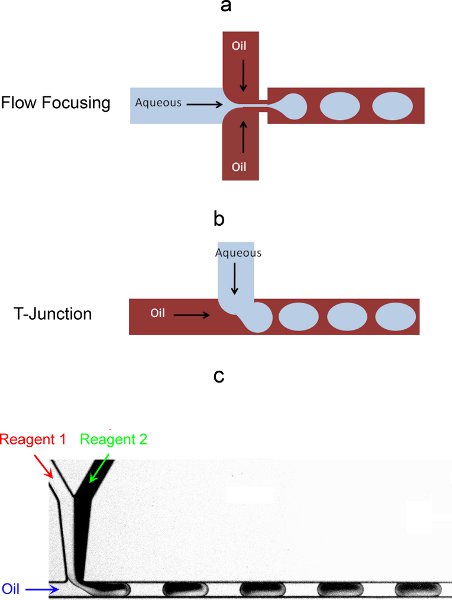
Figure 3. On-chip droplet formation strategies: a) flow-focusing configuration; b) T-junction configuration. c) On chip encapsulation in droplets of two originally separate aqueous reagents.
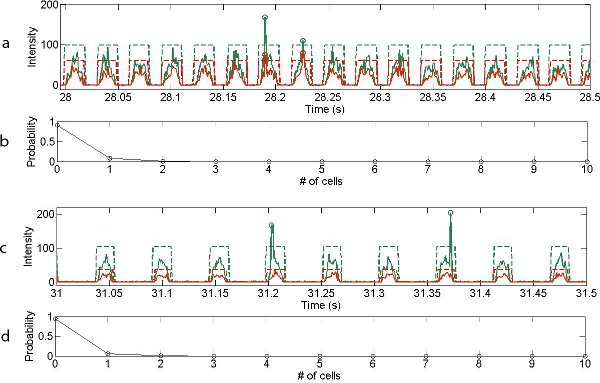
Figure 4. Optical readout of 0.2 s traces recorded for (a) dead and (c) live cells (flow rates for cell suspension: 1 μl min-1, oil: 2 μl min-1). Each arch-shaped signal corresponds to the weakly fluorescent LB medium that forms the aqueous droplet, with droplet boundaries marked with dashed lines. Green signals represent readout under 633nm and red signals over 633 nm wavelengths. Cells are distinguished by the vertical spike arising from the DNA intercalating dyes. Probability distribution for cellular occupancy within single droplets for (b) dead cells and (d) live cells.
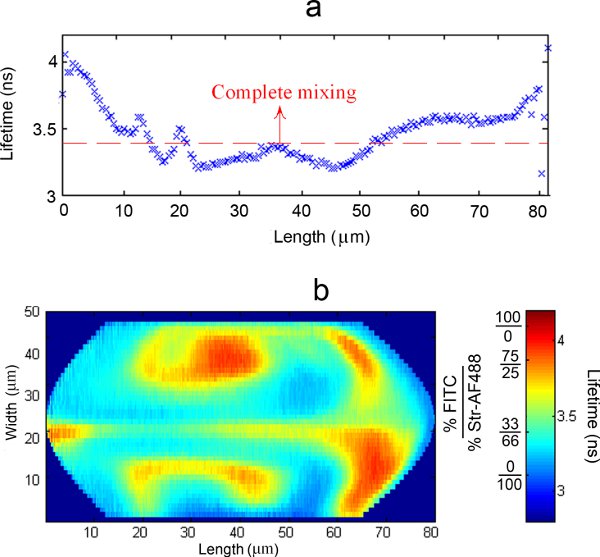
Figure 5. a) Typical averaged lifetime trajectory along the length of a droplet at a particular width; b) Typical representation of the combination of all the averaged trajectories probed along the full width of the droplets into a 2D lifetime (or concentration, expressed as % FITC / % Str-AF488) map.
