To bypass the lethality induced by ubiquitous expression of expanded Atxn3 under the control of da-Gal4, we introduced temporal regulation of Gal4 with Gal80ts. But before proceeding with the collection and freezing of large numbers of flies, we wanted to determine the expression dynamics of our transgene under the control of Gal80ts and da-Gal4. To do this, we generated the crosses, left the progeny at 18 °C, collected the adult flies, and maintained them for 24 hr at 18 °C. Then, we placed the flies at the restrictive temperature (29 °C) and incubated them for 0, 6, 12, 24, 36, 48, and 72 hr. Next, we detected the accumulation of the expanded polyQ allele by western blot from single flies following published protocols10. Expression was first detected, but faint 6 hr after temperature shift and continued to increase until 48 hr, when it reached saturation levels (Figure 2A). Interestingly, a high molecular weight band of insoluble polyQ appeared after 48 hr, signaling the time it takes for the protein to form large aggregates. We also dissected the fly brains at each time point to determine the distribution of the expanded polyQ allele by immunofluorescence (Figure 2B). The protein was first detected 24 hr after the temperature shift, although with uneven distribution. Between 24 and 48 hr the levels of protein continued to rise, making several brain centers clearly visible, including the antennal lobes and the mushroom body projections (Figure 2B). Between 48 and 72 hr the expression of the expanded polyQ allele reached maximal levels, suggesting that the Gal4 system was fully activated at this time. Based on these results, we selected 24 hr after the temperature shift as the first time point (day 1) for subsequent experiments. The collections of flies 10 and 20 days old were also measured starting from the temperature shift (time 0), which is the time when the experiment starts with the new expression of the pathogenic and control transgenes.
Once we verified that Atxn3 was detected after 24 hr under the control of Gal80ts and da-Gal4 at 29 °C, we wondered whether these flies would show signs of neurodegeneration over 20 days. Since these flies start expressing the pathogenic proteins as adults, we feared that they might need a long incubation time to show neurodegenerative phenotypes. To determine the toxicity of Atxn3-78Q in these conditions, we collected flies expressing LacZ, Atxn3-27Q, and Atxn3-78Q, and recorded their longevity. Whereas flies expressing LacZ and Atxn3-27Q displayed over 90% viability after 20 days, flies expressing Atxn3-78Q showed low survival at day 20 (30%) (Figure 3). In fact, Atxn3-78Q flies started to die by day 10 (7% mortality) and by day 15 the loss was significant (20%), which accelerated in the last five days. Thus, these results indicated that adult expression of a pathogenic gene resulted in shortened lifespan, a key sign of the neurodegenerative phenotypes induced by Atxn3-78Q.
One of the most critical aspects of this protocol was the handling and preservation of specimens after freezing to ensure the quality of the material extracted. We extracted between 15-80 μg of total RNA per 200 heads (depending on age and genotype) before DNase treatment according to NanoDrop. Approximately one third (6-25 μg) were recovered as highly pure RNA after DNase treatment by the ratio of absorbance at 260 nm/280 nm. However, we found that the best way to determine the quality of RNA for the preparation of cDNA libraries for RNA-seq was to analyze total RNA on the BioAnalyzer. Fortunately, only a small amount of RNA (5 ng) was used on the BioAnalyzer, so it did not impact the ability to produce several libraries per sample. As an example, we ran two samples of total RNA before DNase treatment on a BioAnalyzer chip, one of them suspected of being degraded (Figure 4A-C). The high quality RNA (sample 1) produced three sharp RNA bands, two just below 2,000 nucleotides (nt) in size and a smaller band below 200 nt, representing the ribosomal RNAs (Figure 4A). The two bands around 2,000 nt correspond to the 18S rRNA (smaller peak) and two fragments from the 28S rRNA (larger peak), which is a unique aspect of rRNA biology in Drosophila (see Figure 4B). These qualities were also observed in the BioAnalyzer traces (Figure 4B). In contrast, a degraded RNA sample (sample 2) did not produce the sharp peaks of the ribosomal RNAs, and accumulated multiple bands of less than 500 nt (Figure 4A). This size distribution was readily distinguished in the BioAnalyzer traces (Figure 4C), in which broad peaks of shorter size were noted. Also important to note was the difference in the units of the y-axis between the two RNA samples, which demonstrated that the degraded sample had less RNA. Only RNA displaying maximum quality (NanoDrop ratio of absorbance at 260 nm/280 nm between 1.8 and 2.1, with 2.1 being optimal, see protocol step 3.8) will be used for the generation of libraries.
For the extraction of metabolites, we followed a classic water / methanol / chloroform (2.0:2.0:1.8) extraction procedure3 split into two steps to maximize extraction of both polar and non-polar metabolites35. After separating the polar and non-polar phases, we reconstituted them in deuterated water or deuterated chloroform, respectively, and added internal standards for analysis by NMR. NMR spectra obtained using this extraction method were well resolved with flat baselines and narrow lines (Figure 5), indicating the purification of high-quality extracts, suitable for both metabolic profiling and the identification of large numbers of metabolites. Figure 5 shows the identification of a few prominent peaks corresponding to highly abundant metabolites, such as glucose and sucrose, and two key metabolic acids, lactate and acetate, according to chemical shifts in the literature7. The chemical shift (in parts per million) represents the electromagnetic shielding of a molecule relative to a standard molecule (TMS, first peak from the right). More shieldedmolecules have higher electron density around the nucleus and will appear to the right of the spectrum, including alkyls. Deshielded molecules such as amide and aromatic hydrogens will appear to the left of the spectrum. The busy middle section (3-4 ppm) is enriched for sugar molecules.
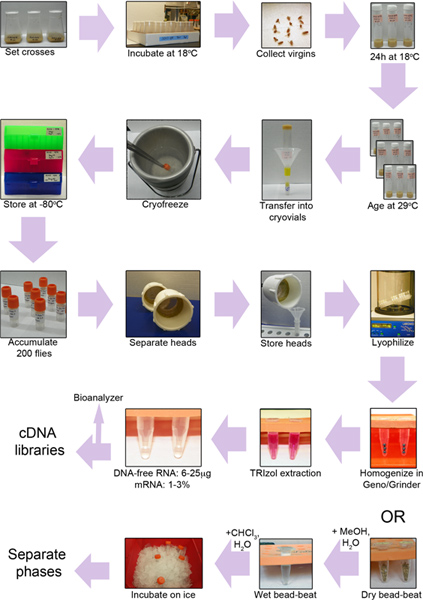
Figure 1. Experimental workflow. First, we set crosses to collect flies of the desired genotypes (top row). Then, we shift virgins to 29 °C to activate gene expression, age them for 1, 10, and 20 days, and flash-freeze them (second row). We next collect frozen heads using a microsieve (third row). The heads are freeze-dried, ground up, and treated for the extraction of RNA (fourth row) or metabolites (bottom row).
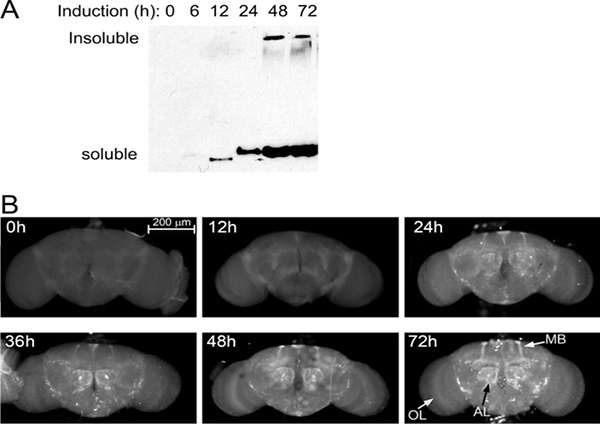
Figure 2. Expression kinetics. (A) Western blot showing Atxn3-78Q expression (Gal80ts; da-Gal4 /UAS-Atxn3-78Q) in flies incubated at 29 °C from 0 to 72 hr. A faint band is first detected at 6 hr and saturation is reached by 48 hr after induction. (B) Immunofluorescence of whole brains in the same flies. Brains were dissected, fixed, and stained with an antibody that recognizes the expanded polyQ protein. The protein is first detected in the mushroom body (MB) projections and the antennal lobes (AL). Between 48 and 72 hr, the expression reaches maximum levels and is highly visible in brain centers with abundant innervation. The expression level per neuron is weak as seen in the optical lobe (OL), except in a few large neurons between the OL and the central brain.
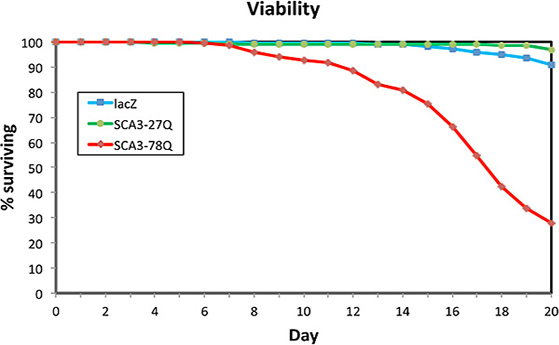
Figure 3. Atxn3-78Q reduces lifespan. Flies expressing LacZ, Atxn3-27Q, and Atxn3-78Q ubiquitously in adult stages (Gal80ts; da-Gal4) were monitored for their longevity at 29 °C. Flies expressing LacZ (blue) and Atxn3-27Q (green) showed low levels of mortality by day 20. In contrast, flies expressing Atxn3-78Q (red) displayed a pronounced mortality starting at day 10. By day 15 they showed a 20% reduction in viability, which accelerated over the next five days, reaching a 70% mortality by day 20.
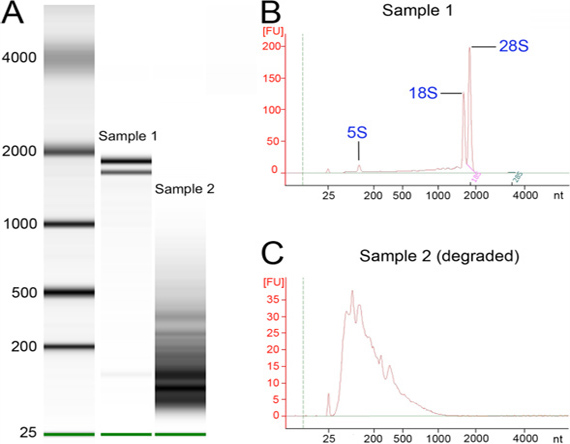
Figure 4. RNA quality assessment. RNA from high quality and degraded samples was run on a BioAnalyzer chip. (A) BioAnalyzer run with a high quality RNA (sample 1) and a degraded RNA (sample 2) next to a size marker (left lane). Note the three sharp peaks corresponding to ribosomal RNA in the central lane. (B) BioAnalyzer tracing for sample 1, with two characteristic strong peaks just below 2,000 nt and another below 200. (C) BioAnalyzer tracing for sample 2, which consists of mostly degraded RNA. Note the difference in units on the y-axis between panels B and C.
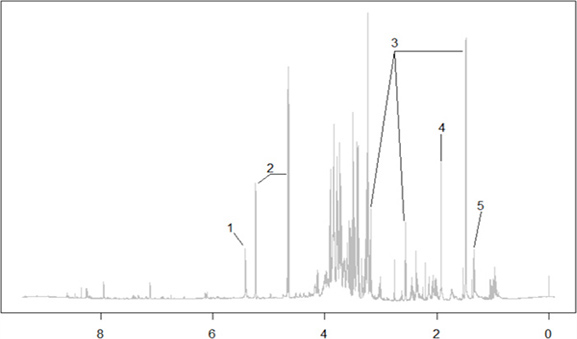
Figure 5. Representative 1D NMR spectra of polar metabolites. Representative 1D proton NMR spectrum of polar metabolites from Drosophila and complementary 2D COSY (correlation spectroscopy) and HSQC (heteronuclear single quantum correlation) experiments for peak assignment. Metabolites identified according to known chemical shifts- 1: Sucrose; 2: Glucose; 3: Beta-alanine; 4: Acetate; 5: Lactate.
