Flow cytometry is a useful tool to identify and characterize different cells and it is the technique of choice to identify specific immune cells, such as monocytes, granulocytes, T cells, B cells, natural killer (NK) cells, dendritic cells (DCs), and other subpopulations of leukocytes.
In the effort to better characterize white blood cells behavior in response to nanoparticles, we performed internalization assays with primary leukocytes isolated from the blood of healthy donors (Figures 1 and 2) and with human monocyte cell line (THP-1 cells, Figure 3).
As reported in Figure 1A, the three major blood leukocyte subpopulations were clearly identified by forward and side scattering after PBMCs isolation. Moreover, after FITC-SiO2 treatment, lymphocytes (grey), monocytes (blue) and granulocytes (red) have a different nanoparticle internalization rate as shown by green fluorescence intensity (Figure 1B). The described protocol allows the purification of primary CD14 positive monocytes from PBMCs. Figure 2A reports the flow cytometry dot plot of CD14+ monocytes in presence of FITC-SiO2. Figure 2B shows FITC-SiO2 internalization quantified in the same cells and expressed in logarithmic scale histogram.
Similar internalization experiments were performed on THP-1 monocytes treated with increasing concentrations of FITC-SiO2 nanoparticles. Untreated cells were used as negative control. Figure 3A shows a dose-dependent increase in side scattering with an unchanged forward scattering in THP-1 cell line. In Figure 3B, together with the histograms of SSC and FSC at each FITC-SiO2 nanoparticle concentration tested, mean fluorescence intensity (MFI) quantification is presented. This data suggest that treatment with FITC-SiO2 nanoparticles induces a dose-dependent internalization in monocytes highlighted by the enhancement of intracellular granularity (side scattering) and fluorescence (green channel).
To gain further insights into the type of interaction between immune cells and nanoparticles, primary mixed glial cultures were isolated and microglia, the central nervous system resident immune cells, was purified. The use of a transgenic mouse model expressing green fluorescent microglia permits the visualization of different neuro-inflammatory mechanisms. The transgenic mouse B6.129P-CX3CR1tm1Litt/J used in this work expresses the green fluorescent protein (GFP) under the control of CX3CR1 promoter12. After 7 days in vitro (DIV), fluorescence microscopy shows a mixed primary glial culture with a large number of astrocytes (GFP negative adherent cells) and some green cells (GFP positive, Figure 5A). In this mouse model, three glial subpopulations can be distinguished by flow cytometry with a single CD11b-antibody staining: the first CD11b–GFP– (astrocytes and other glial cells), a second distinct group of microglial CD11b+GFP+ cells, and a third CD11b+GFP– subpopulation (Figure 4A). These two latter subpopulations are both able to internalize nanoparticles with a slight increased efficiency by the GFP+ population (representing the patrolling immature microglia by the transcription of CX3CR1 promoter), as shown by flow cytometry analysis (Figure 4B). The occurred internalization can be further verified by confocal microscopy using the same final concentration of Rhodamine-SiO2 nanoparticles as shown in Figure 5B.
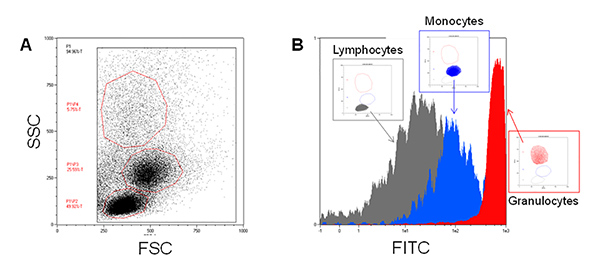
Figure 1. FITC-SiO2 nanoparticle internalization in isolated blood leukocytes. A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of Ficoll-Paque isolated blood leukocytes. B) Green fluorescence overlay histogram plot of the three major blood leukocyte cell subpopulations in presence of 1 nM FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
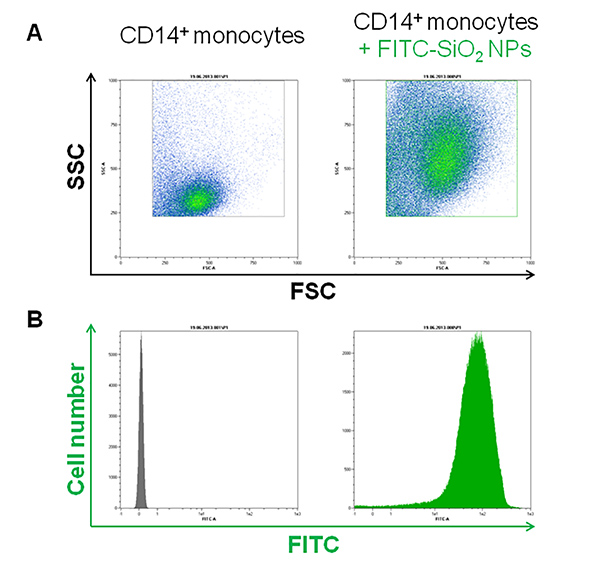
Figure 2. FITC-SiO2 nanoparticle internalization in CD14+ purified monocytes. A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of purified CD14 positive monocytes. B) Green fluorescence histogram plot of the purified monocyte subpopulation in presence of 1nM FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
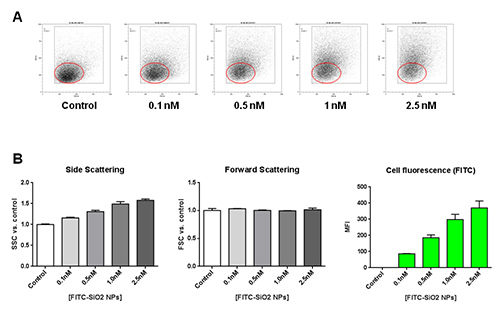
Figure 3. Effects of FITC-SiO2 nanoparticle internalization on THP-1 cells. A) Representative forward scattering (FSC) versus side scattering (SSC) flow cytometry dot plot of THP-1 monocyte cell line, following 1 hr exposure of FITC-SiO2 nanoparticles increasing concentration. B) Concentration-dependent variation of the side scattering (SSC), forward scattering (FSC) and green fluorescence in presence of FITC-SiO2 nanoparticles (+45 mV) for 1 hr. Please click here to view a larger version of this figure.
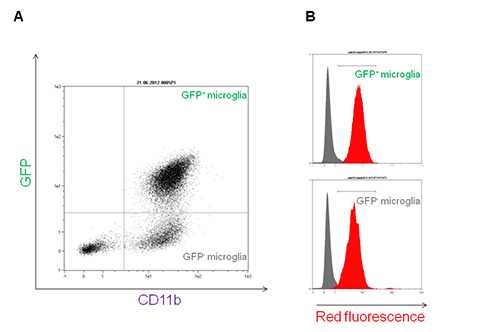
Figure 4. Rhodamine-SiO2 nanoparticle internalization into primary microglia isolated from B6.129P-CX3CR1tm1Litt/J mice. A) Green Fluorescent protein (GFP) versus CD11b-VioBlue flow cytometry dot plot of primary mixed glia isolated from B6.129P-CX3CR1tm1Litt/J mice. CD11b+GFP+ and CD11b+GFP– populations are shown in the upper right and lower right quadrants, respectively. B) Red fluorescence overlay histogram plot of microglia subpopulations in presence of 1 nM Rhodamine-SiO2 nanoparticles (+45 mV) for 30 min (red histogram) versus control (grey histogram). Please click here to view a larger version of this figure.
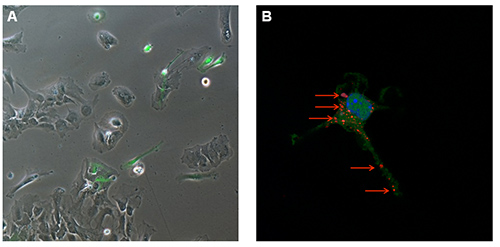
Figure 5. Visualization of GFP+-microglia. (A) Fluorescence microscopy at 7 DIV and (B) Confocal microscopy of Rhodamine-SiO2 nanoparticle internalization (red arrows) in GFP+-microglia.Please click here to view a larger version of this figure.
