At present, there are many instances in the field of neurobiology and neurovirology where undifferentiated SH-SY5Y cells are being used as a functional model for human neurons27-36, and importantly, undifferentiated cells may lack phenotypes such as optimal viral uptake2 that are necessary for accurate interpretation. It is critical that when using SH-SY5Y cells or any other in vitro neuronal system, cells are appropriately differentiated into neurons, in order to obtain data that is the best possible representation of what may be occurring in neurons in vivo. The above protocol yields highly viable, homogenous, differentiated neuronal cultures within 18 days, that can be used for subsequent biochemical and imaging analyses. Undifferentiated SH-SY5Y neuroblastoma cells demonstrate a large, flat, epithelial-like phenotype with numerous short processes extending outward (Figure 2A), while differentiated cells possess several neuritic projections that connect to surrounding cells (Figure 2B). It is important that when differentiating SH-SY5Y cells, cells are incubated with trypsin for a minimal length of time to ensure that only neurons are released from the dish. This leaves behind the undifferentiated epithelial cells that would otherwise contaminate the differentiated neuronal cell population. The neuronal characteristics of fully differentiated SH-SY5Y cells are demonstrated by techniques such as immunofluorescence detection of classical neuronal markers (Figure 3).
SH-SY5Y cells demonstrate a variety of different phenotypes during the course of differentiation and it is important to be able to identify healthy neurons from those that are stressed. On differentiation day 1, prior to the introduction of Differentiation Media #1, cells have a flat, retracted phenotype with short, stubby processes (Figure 4A). Following 5 days of serum deprivation, SH-SY5Y cells begin to develop longer projections and demonstrate a more neuronal phenotype (Figure 4B). Passaging is a harsh process for SH-SY5Y cells, and on days immediately following passaging, cells appear unhealthy. This is evidenced by cell body clumping and the presence of fewer, shorter processes. (See before images: Figures 4C and 4E, and after images: Figures 4D and 4F). Approximately 48 hr following a split, cells appear to recover, and fully mature, differentiated neurons are obtained at Day 18 (Figure 2B). This is evidenced by a reduction in cell body clumping, and extension of numerous thin, branched neuritic processes that often connect to neighboring cells.
Factors that are essential to obtaining reproducible and viable neuronal cultures include using heat-inactivated FBS, minimizing trypsin incubation time, and gentle trituration. Importantly, this protocol details the use of four different media formulations with various concentrations of hiFBS, thereby creating a gentle transition into a serum-starved state for the cells. When mature SH-SY5Y cells are healthy, they demonstrate numerous projections that connect to surrounding neurons (Figure 5A). It should be noted that a distinctive epithelial phenotype will overtake the neuronal cultures if they are mishandled during the differentiation process (Figure 5B). Additionally, if neurons begin to die, neurites will retract, cell bodies will begin to clump together and round up, and debris from neurite degeneration will start to accumulate at and around cell processes. This process is akin to what is demonstrated in Figures 5C and 5D. While Figure 5C shows a more natural progression of cell death following nutrient and environmental deprivation, Figure 5D shows differentiated SH-SY5Y neurons 4 hr following infection with an HSV-1 strain, KOS, at a multiplicity of infection (MOI) of 106 PFU. This protocol has been thoroughly optimized in the lab and yields reproducible, homogeneous populations of neurons, which are essential for downstream analyses and experimentation.
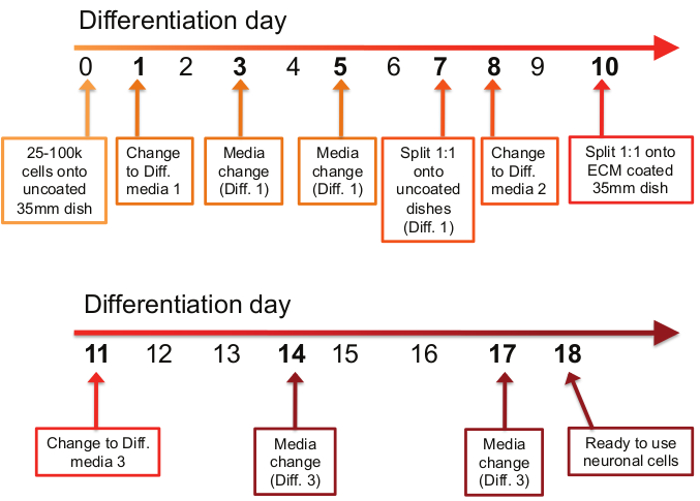
Figure 1: Timetable of differentiation procedure. The differentiation process consists of 11 steps spread out over the course of an 18 day period. On the first day of the differentiation protocol (day 0), between 25,000 and 100,000 cells are plated onto uncoated 35 mm dishes. On days 1, 3, and 5, old media is removed and Differentiation Media #1 is applied. On day 7, cells are split 1:1 onto uncoated 35mm dishes in Differentiation Media #1. On day 8, the media is changed to Differentiation Media #2, and on day 10, cells are again split 1:1, but this time onto ECM-coated 35 mm dishes in Differentiation Media #2. On days 11, 14, and 17, old media is removed and Differentiation Media #3 is applied. On day 18, differentiated neurons are ready to use for downstream applications.
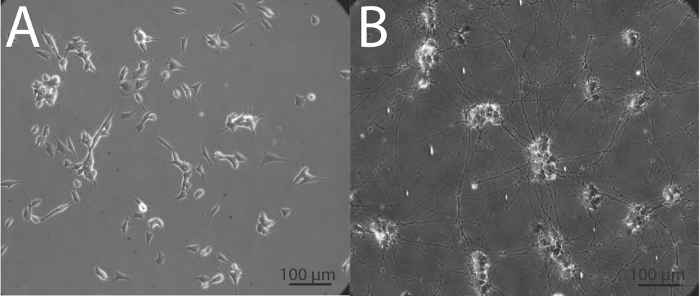
Figure 2: Morphological appearance of undifferentiated and differentiated SH-SY5Y cells. (A) Undifferentiated SH-SY5Y cells have a flat phenotype with few projections while (B) differentiated SH-SY5Y neurons demonstrate extensive and elongated neuritic projections. Images were obtained in phase at 20X magnification using an inverted epifluorescence microscope.
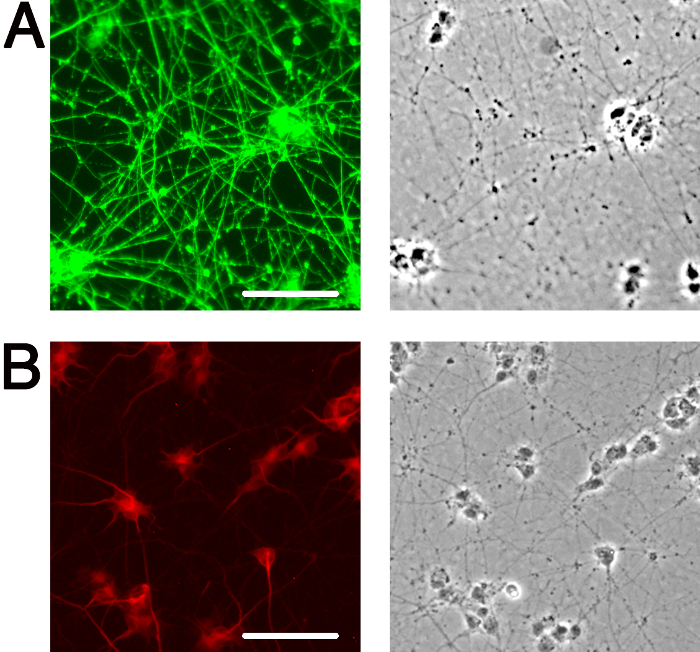
Figure 3: Markers of neuronal differentiation. Immunofluorescence illuminates neuronal features of fully differentiated SH-SY5Y cells. (A) Anti-SMI31 (green) stains the phosphorylated neurofilament H in the extensive network of neurites. (B) Anti-MAP2 (red) labels microtubule-associated protein 2, revealing the neuronal soma and proximal portion of neurites. Corresponding phase image for each immunofluorescence panel is shown to the right. Images were obtained at 10X magnification using an inverted epifluorescence microscope. Scale bar, 100 μm.
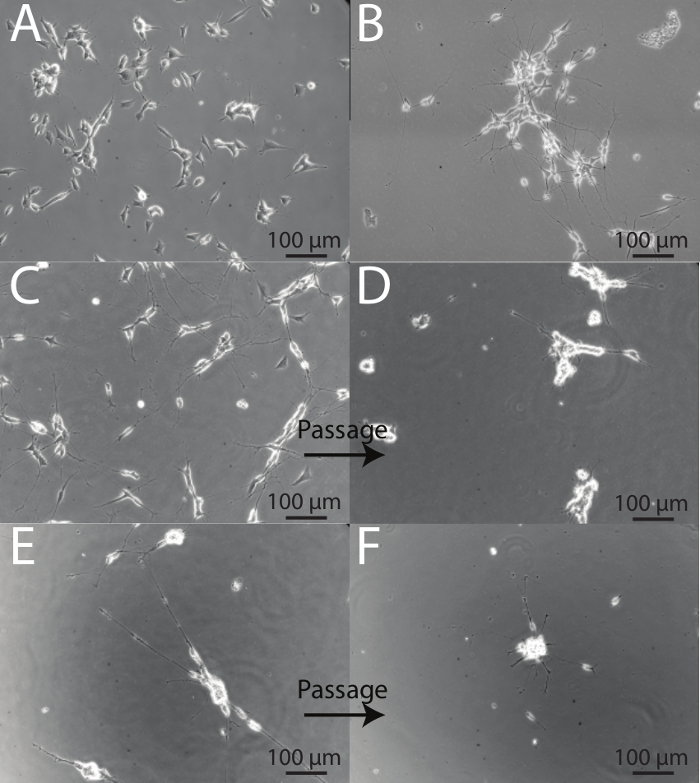
Figure 4: Intermediate steps of the differentiation protocol. (A) Day 1 of differentiation. Cells maintain a retracted phenotype with short projections. (B) Day 5 of differentiation. Cells have been exposed to 5 days of serum deprivation (Differentiation Media #1). The surviving cells begin to elongate and form longer processes that connect with neighboring cells. (C) Day 7 of differentiation prior to split. Cells demonstrate high numbers of long processes, with fewer numbers of cells demonstrating an epithelial-like phenotype. (D) Day 8 of differentiation, one day after the first passage. Following splitting, cell bodies form clumps and processes appear short as a result of the passaging procedure. (E) Day 10 of differentiation, prior to split onto ECM-coated plates. Cells demonstrate longer processes that make connections with nearby cells. Cell body clustering is also evident. (F) Day 11 of differentiation, one day following second split. Cells are stressed following the second passage and many neurons are ultimately lost. However, the remaining population is viable, homogenous, and neuronal in phenotype. Cell bodies produce larger clusters and processes begin to emit from the base of the clusters.
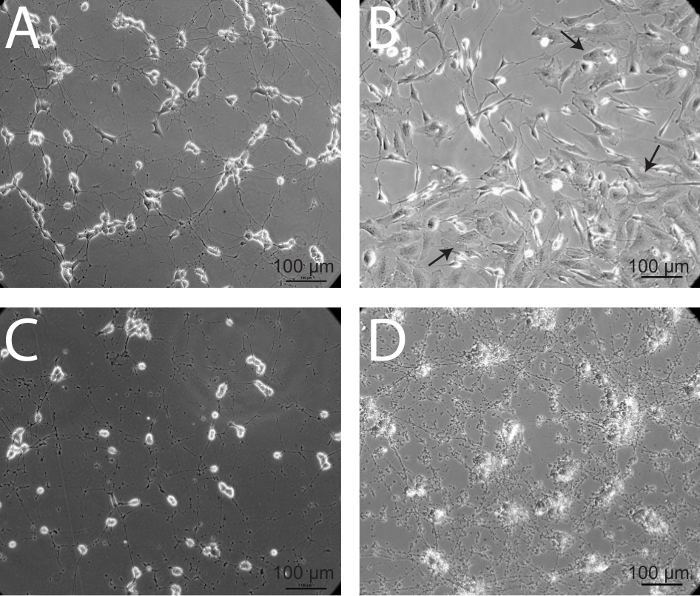
Figure 5: Epithelial overgrowth and neuronal death – two alternative outcomes of the differentiation process. (A) Healthy, mature SH-SY5Y neurons demonstrate diffuse axonal projections connecting to neighboring cells. (B) In some instances, cells with a more epithelial-like phenotype overtake maintenance cultures. This over-population of epithelial-like cells may be due to infrequent passaging of maintenance cultures. These cultures should be discarded, as the epithelial-like cells will continue to outnumber neurons. Arrows denote epithelial-like cells. (C) When SH-SY5Y cells are unhealthy and begin to die, cell bodies round up and processes degrade, generating a significant amount of debris. (D) Noticeable accumulation of cellular debris and retraction of neuronal processes is evident in mature, differentiated neurons, 4 hr following infection with a strain of herpes simplex virus 1 (HSV-1) KOS at a multiplicity of infection (MOI) of 106 PFU.
| Component | Details | Stock | Instructions | ||||
| 10 μM RA | All-trans retinoic acid | 5 mM | Resuspend 50 mg RA in 33.3 ml 95% EtOH. RA is sensitive to heat, light, and air. Keep in a dark bottle and store at 4 °C for up to 6 weeks. Use at 1:500 dilution and dilute into differentiation media immediately prior to use | ||||
| (300.44 g/mol) | |||||||
| 1x B-27 | B-27 Supplement | 50x | Thaw 1-10 ml bottle and aliquot remainder into single-use 1 ml aliquots and store at -80 °C. Store 10 ml bottles at -20 °C | ||||
| 20 mM KCl | Potassium Chloride | 1 M | Add 250 ml water to 18.6 g KCl and sterile filter. Store at room temperature | ||||
| (74.55 g/mol) | |||||||
| 2 mM db-cAMP | dibutyryl cyclic AMP | 1 M | Resuspend full bottle by adding 2.04 ml water to 1 g db-cAMP. Sensitive to light and moisture. Store in aliquots of 100 μl or 200 μl at either -20 °C or -80 °C | ||||
| (491.37 g/mol) | |||||||
| 50 ng/ml BDNF | Brain-derived neurotrophic factor (BDNF) | – | Centrifuge vial to get powder to bottom. Resuspend 10 μg vial in 1 ml Neurobasal + 1x B27 or 5 μg vial in 0.5 ml Neurobasal + 1x B27 to get 10 μg/ml. Use at 1:200 dilution. Store working aliquots at -80 °C (ex: 250 μl) | ||||
| hiFBS | Heat-inactivated Fetal Bovine Serum | – | Aliquot thawed FBS into 50 ml conical tubes. Heat at 56 °C in a water bath for 30 min. Remove and freeze working aliquots at -20 °C | ||||
Table 1: Stock solutions and components.
| Basic Growth Media | ||
| Component | Volume for 500 ml | Dilution |
| EMEM | 415 ml EMEM | |
| 15% hiFBS | 75 ml hiFBS | |
| 1x Pen/Strep | 5 ml Pen/Strep | 1:100 |
| 2 mM Glutamine | 5 ml Glutamine | 1:100 |
| *Keep for 6 weeks maximum | ||
| Differentiation Media #1 | ||
| Component | Volume for 50 ml | Dilution |
| EMEM | 48 ml EMEM | |
| 2.5% hiFBS | 1.3 ml hiFBS | |
| 1x Pen/Strep | 500 μl Pen/Strep | 1:100 |
| 2 mM Glutamine | 500 μl Glutamine | 1:100 |
| 10 μM RA | 100 μl RA (5mM stock) | 1:500 |
| *Keep for 2 weeks maximum and add RA immediately prior to use | ||
| *Do not keep extra media once RA is added – RA is unstable | ||
| Differentiation Media #2 | ||
| Component | Volume for 50 ml | Dilution |
| EMEM | 49 ml EMEM | |
| 1% hiFBS | 500 μl hiFBS | |
| 1x Pen/Strep | 500 μl Pen/Strep | 1:100 |
| 2 mM Glutamine | 500 μl Glutamine | 1:100 |
| 10 μM RA | 100 μl RA (5mM stock) | 1:500 |
| *Keep for 2 weeks maximum and add RA immediately prior to use | ||
| *Do not keep extra media once RA is added – RA is unstable | ||
| Differentiation Media #3 | ||
| Component | Volume for 50 ml | Dilution |
| Neurobasal | 47 ml Neurobasal | |
| 1x B-27 | 1 ml B-27 (50X stock) | 1:50 |
| 20 mM KCl | 1 ml KCl (1M stock) | 1:50 |
| 1x Pen/Strep | 500 μl Pen/Strep | 1:100 |
| 2 mM GlutamaxI | 500 μl GlutamaxI (100x stock) | 1:100 |
| 50 ng/ml BDNF | 250 μl BDNF stock (10 μg/ml) | 1:200 |
| 2 mM dibutyryl cyclic AMP (db-cAMP) | 100 μl db-cAMP (1M stock) | 1:500 |
| 10 μM RA | 100 μl RA (5 mM stock) | 1:500 |
| *Keep for 2 weeks maximum and add RA immediately prior to use | ||
| *Do not keep extra media once RA is added – RA is unstable | ||
Table 2: Media recipes.
