1. Sample Preparation: Melt Filling of SWNTs with Mercury Telluride (HgTe) and Other Materials
Caution: Some chemicals used in this protocol can be hazardous to health. Please consult relevant material safety data sheets before any chemistry takes place. Utilize appropriate personal protective equipment (lab coat, safety glasses, etc.) and engineering controls (e.g., glove box, fume hood etc.) when handling carbon nanotubes and mercury telluride.
- Preheat ~50 mg SWNTs to 400 °C in dry air or 500 °C in vacuo.
NOTE: If dry air is used, there may be some loss of volume of the SWNTs, typically 20-40%. Preheating of SWNTs prior to filling in this way has been reported to improve the proportion of SWNTs filled with molecules or crystals12. - In an Argon glove box grind ~20 mg of the preheated SWNTs26 with an equal volume of the filling material (in this case mercury telluride) for >20 min using an agate mortar and pestle, applying force to produce an intimate mixture.
- While still in the glove box, transfer the whole amount of the SWNT/filling material mixture to an 8-10 mm (internal diameter), 6-10 cm long silica quartz ampoule sealed at one end and open at the other.
NOTE: A filter paper funnel is useful in 1.3. Do not use ordinary lab glass ampoules as this grade of glass may melt and damage the furnace. - Inside the glove box temporarily seal the open end of the ampoule with plastic film for transfer to the vacuum line. Remove the film once it is secured to a vacuum line.
2. Evacuation and Filling Step
- Seal the ampoule containing the SWNT/filling composite under moderate vacuum (typically ~0.1 Pa).
- In a programmable tube or muffle furnace, heat the sealed ampoules at a ramp rate of ~5 °C min-1 to ~100 °C >melting point (m.p.) of the filling with thermal cycling at temperatures of +/- 100 °C for 12 hr periods for a total time of 48 hr.
- Cool the ampoule in the furnace to room temperature at a rate no greater than 5 °C min-1 prior to breaking open and storage.
Hazard: Breaking open ampoules can potentially cause cuts or can implode, presenting a significant eye impact hazard. Eye protection and safety gloves should be worn during this operation. Ampoules may be safely broken by first scoring the ampoule in the center with a glass scribe and then breaking at this position by wrapping in a stiff cloth and then simultaneously applying bending pressure either side of the score point.
3. Sample Cleaning
- Remove exterior compounds to the SWNTs by gently refluxing the sample in a 1:1 mixture of concentrated HCl and HNO3 (total volume 50 ml) for ~1 hr.
- Wash the sample with de-ionized water and filter using a controlled pore filter that has a pore size of ~0.22 µm.
4. Analysis of the Sample by High Resolution TEM (HRTEM)
- Disperse ~ 5 mg of sample in ~5 ml of ethanol with sonication at about 20% of 750 W of power in a tip sonicator with 2 sec on/off pulsing.
- Place 1-2 drops of the dispersion onto 3.05 mm lacey carbon coated HRTEM specimen grids.
- Perform HRTEM27,29 imaging in an 80 kV aberration-corrected HRTEM and equipped with a 4,008 x 2,672 pixel charge-coupled device (CCD).
- Precalibrate the CCD at the same magnification to be used for imaging the nanowire/carbon nanotube composite using (111) lattice planes (separated by 0.235 nm) of dispersed Au nanoparticles.
- Obtain images of discrete filled SWNT samples at a magnification of at least 600,000 times and under optimum Scherzer defocus imaging conditions.
- Confirm the composition of the fillings by energy dispersive X-ray microanalysis (EDX)28,29 ideally using the same HRTEM as used for imaging. Use a ~1 nm electron probe focused onto filled bundles of SWNTs.
5. Confirming the Microstructure of SWNT Embedded Extreme Nanowire by HRTEM Image Simulation
NOTE: For image simulations a standard multislice image simulation package such as SimulaTEM may be used which produces bitmap image (*.bmp) simulations that may be directly compared. For exact details on operation of software through various platforms, please follow manufacturer's protocol.
- Choose a HRTEM image of a nanowire free of drift and with clearly resolved atom columns visible for a distance along the SWNT of ~5 nm. The long axis of the nanowire/nanotube composite should be orthogonal with respect to the electron beam.
- Generate atomic coordinates by plotting atom positions directly onto atom columns using a calibrated image (step 4.4).8 in the case of a high crystal symmetry structure; a single image is required11. Add 2-3 atomic layers in projection to the model to complete the structure. For a low symmetry model, resolve the crystal structure from two or more different projections of two or more crystallographically identical fragments in two separate nanotubes10.
- Generate *.xyz nanotube coordinates for a nanotube of suitable diameter using a suitable program (e.g. TubeGen 3.4) determined according to the relation.
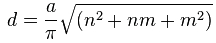
Where d is the diameter, a is the C-C bond distance (0.246 nm) and n and m the tube conformation. The nanotube should be large enough to accommodate the outside volume of the crystal generated from step 5.2 taking into account the van der Waals radius of the carbon atoms of the nanotube wall (0.17 nm). - Assemble composite atomic coordinates from the nanowire/nanotube composite using a suitable structure manipulation program (e.g. Crystalmaker) such that the 1D crystal is inserted into and aligned along the common central axis of the nanotube and then save the final model coordinates in *.xyx or *.pdb format.
- Produce image simulations of nanowire/nanotube composite using a standard multislice simulation protocol (e.g. SimulaTEM) using the atomic coordinates generated in 5.3.
- Simulate the composite in a preliminary orientation such that the long axis of the composite is orthogonal to the electron beam. Use simulation parameters consistent with the accelerating voltage (e.g. 80 kV) and a coefficient of spherical aberration (Cs; e.g. 0.001 mm) consistent with the HRTEM instrument.
- Visually compare the appearance of the simulation to the image. If a good visual match is not obtained rotate the fragment about the long axis of the extreme nanowire/nanotube composite by a suitable interval (e.g. 10°) and then re-simulate. Repeat this step for a full 180° rotation of the composite.
- If it is suspected that the experimentally imaged fragment is tilted with respect to an ideal orthogonal orientation, repeat steps 5.5 to 5.7 with a tilt of ±10° until a good match is obtained.
6. Preparation of Sample Suitable for Raman Spectroscopy
Hazard: The sonication of nanotube solutions may be able to form an aerosol containing tubes or filled tubes and if the samples are subsequently not handled correctly this could lead to the operator breathing in nanotubes or filled nanotubes.
- Place 20 mg of nanotube product into a vial, add 20 ml ethanol and seal lid.
- Place vial in bath sonicator and sonicate at 20 W for ~20 min until liquid turns dark. Leave for ~5 hr to allow nanotube aerosolized in vial to settle out.
- Gently swirl vial to agitate bottom suspension if present.
- Using a pipette, drop coat 10-20 µl of suspension onto oxide coated silicon substrate (5 mm x 10 mm) and allow ethanol to evaporate.
7. Mounting Sample in Cryostat
- Place a droplet of metallic conductive paint (approximately 2 mm2) on the Cryostat cold finger and gently place silicon sample on paint droplet and let dry for ~2 hr.
- Seal cryostat as per manufacturer's protocol and bolt Cryostat onto XYZ stage and pump Cryostat to 10-6 mbar using an oil free pump.
8. Initial Setup and Optimization of Raman System
NOTE: Please refer to the experimental schematic presented in Figure 10 before reading the following sections of the protocol.
- Set incident wavelength to desired value (e.g. 800 nm) using a tunable laser source as per manufacturer's protocol.
- Tune laser pre-filter (C in Figure 10) as per manufacturer's protocol to maximize transmission of laser power through filter. If using a Volume Bragg Grating (VBG) modify the optical set up in accordance with Figure 10 and use the following sub procedure.
- Rotate VBG around vertical axis to reduce transmission of laser through the VBG. Fine tune using VBG mirror mount.
- Position mirror into Bragg reflected beam and retroreflect beam back onto VBG. Adjust mirror to suppress transmission of the retroreflected beam through VBG.
- Measure laser power transmitted through iris 1 and fine adjust VBG and retroreflecting mirror to maximize transmission of laser power.
- Adjust the post-filter (C) mirrors (M1 and M2) to return laser beam to predefined path by repositioning reflections from relevant beam splitters (BP1 and BP2) onto the two beam observation cameras (C1 and C2).
- Measure laser photon energy by indirectly scattering into spectrometer. Do not scatter light directly into the spectrometer as this can damage sensitive spectroscopy cameras.
- Adjust half wave plate (HWP1) to set power incident upon objective (and PM2) to ~1 mW.
- Using the imaging optics (Figure 10: blue dashed line), check sample image and ensure laser spot is in desired location with no stigmation (optimize with 8.3 if there is).
NOTE: The next 10 steps are used initially to ensure laser is efficiently coupled into spectrometer. Once complete, these steps do not need repeating until a significant change is made in the optical setup. - Adjust sample position as per 8.6 so that laser spot is focused on a clean area of silicon.
- Set spectrometer to zeroth order and use observation camera built in to spectrometer (Figure 10 component E) to view image of the input slit on first stage of spectrometer (Slit 1).
- Open Slit 1 and check, using the observation camera, that the reflected laser light from the sample is entering slits.
- By adjusting coupling lens (L3) ensure center laser spot is on input slits horizontally and on camera vertically.
- Repeatedly reduce Slit 1 width and repeat 8.10 to ensure that laser properly centered on Slit 1 and camera. During this process adjust focusing of coupling lens to minimize scatter of laser light from Slit 1 ensuring that reflected laser light is focused approximately at the plane of Slit 1.
- Open Slit 1 so that the reflected laser light is not significantly clipped by Slit 1.
- Set up spectrometer software, as per manufacturer's protocol, to collect Raman scattering from 520 cm-1 silicon Raman peak.
- Set power to 10 mW.
- Take repeated Raman spectra with one second exposures to begin focusing.
- Adjust Z-focus of sample until well-defined 520 cm-1 Si peak is observed.
- Maximize this signal by adjusting input half-wave plate (HWP2) (this will depend on the polarization of the outgoing resonance and the gratings in the spectrometer), input lens and Z-focus of sample.
9. Measurement of Single Raman Spectrum
- Set desired temperature (4 K) and allow system to equilibrate (~40 min).
- Set incident laser power upon PM2 to ~2 mW.
- Set wavelength on spectrometer software to that determined in step 8.4.
- Set center shift on spectrometer software to 0 cm-1 and acquire a short (500 msec) Raman spectra to measure laser line.
- Use value determined in 9.4 to set new, more precise wavelength in spectrometer software.
- Set center shift and spectral window in software to observe predicted Raman modes and so that the spectral window does not encompass the laser line tail.
- Move sample position to an area of interest (choice of sample position for bundled tubes is important, see section 14 for further details).
- Acquire Raman spectra with CCD focusing on 1 sec exposures as per the manufacturer's protocol.
- Adjust the Z-focus position of the sample using stage controllers to maximize the reflected power at the power meter (PM1).
- Acquire a Raman spectrum using suitable exposure time to get sufficient signal (>1,000 counts absolute).
10. Measurement of Laser Power Dependence of Raman Cross-section
- Set laser wavelength to estimated peak of resonance for specific sample at the working temperature.
- Set a low power (~0.1 mW) and acquire Raman spectra as per section 9.3-9.10 of this protocol.
- Increase power (by ~0.1 mW), ensuring that as HWP1 is rotated the laser spot on sample does not shift. If the laser spot shifts on rotation of HWP1 then realign as per 8.3 to ensure this does not happen and start experiments again.
- Repeat 10.3 for a suitable range of powers to up to ~5 mW. Repeat this process for each working temperature.
- Plot the fitted intensity of observed Raman features versus laser power and determine the linear region. Perform all subsequent Resonance Raman experiments at that temperature with 80% of the upper power limit of the linear region.
11. Measurement of Laser Energy Dependence of Raman Cross-section
- Set desired excitation wavelength (e.g. 700 nm) and align system accordance with sections 8.1-8.6, setting incident power to that determined from section 10.
- Maximize reflected power by adjusting Z-focus of sample and continuously acquire 1 sec exposures in order to check the intensity of the main Raman features.
- Set exposure time and accumulations to maximize signal on CCD without saturating detector.
- Save spectra noting the in the filename the grating used, exposure time, accumulations, center Raman shift, excitation energy and incident power.
- Set next excitation wavelength by repeating steps 8.1-8.6 ensuring the incident laser power is constant and then maximize reflected power as per step 11.2. For initial results use excitation laser wavelengths approximately 10 nm apart. For presentation quality results laser wavelengths separations of approximately 2 nm are preferable.
- After each six new excitation wavelengths return to one previous excitation wavelength (e.g. 700 nm) and repeat 11.1-11.5. Plot the fitted intensity of one key Raman feature from the repeated spectra as a function of the experimental time and ensure there is no long-term drift.
12. Measurement of Polarization Dependence of Raman Spectra
- Place the polarization analyzer (Pol2) into the path between the objective and the spectrometer as shown in Figure 10 (purple dotted line). Additionally place a half wave plate and polarizer (HWP3 and Pol3) into the optical path prior to the objective; this will be used to rotate the incident polarization. Ensure that all polarizers and HWPs are aligned correctly, so the incident radiation is perpendicular to the optical axis and travels through the center of the optics.
- Adjust Pol3 to pass vertically polarized light. Using a strong Raman signal, e.g. a piece of bare silicon, adjust (Pol2) to pass vertical polarized light and adjust HWP2 to maximize intensity of Raman signal. Note the rotation of the HWP2 and the direction of the polarization of the light going into the spectrometer. Ensure that in the rest of the experiment, the polarization of the light incidence on the experiment is unchanged by adjusting HWP2 to compensate any change in the polarization analyzers pass direction.
- Check the alignment of the polarization analyzer; rotate the polarizer 180° to make sure the signal is still the same.
- Perform step 12.3 with the HWP (rotating it in 90° steps around its full revolution).
- Note the change in signal if any at each step.
- Maximize the reflected power by adjusting the Z-focus and acquire Raman spectra using appropriate input power and exposure time/accumulations.
- Adjust collection polarizer with an appropriate pitch (10°) and adjust HWP in a manner as to maintain the polarization of the light incidence on the spectrometer constant as discussed in 12.2.
- Repeat step 12.7 until the full range of the polarizer has been measured.
- Change the polarization of the light incident onto the objective to horizontal.
- Check that this has not caused the laser spot to move on the sample. Repeat the measurement in steps 12.7 and 12.8. Other incident polarizations may also be used but at least two, perpendicular polarizations are necessary. If using non-horizontal or vertically polarized light the effect of the optics between Pol3 and the objective on the polarization of the light needs to be taken into consideration.
- Check the polarization dependence against that expected for ensembles of 1D objects to determine which Raman features are associated with the encapsulated nanowires (see Representative Results).
13. Measurement of Temperature Dependence of Raman Spectrum
- Set new sample temperature on cryostat controller in accordance with manufacturer's protocol.
- By observing cryostat temperature and the movement of the sample ensure that the cryostat is in thermal equilibrium and there is no sample movement before continuing.
- Correct for any thermal drift of sample position by observing the sample in cryostat with and without laser illumination.
- Ensure that the laser power is in the linear regime following steps from section 10.
- Measure the laser energy dependence of the Raman Cross-section as in section 10. It is common for the optical resonances of the sample to shift with temperature although often the energy of the resonance changes relatively slowly with temperature.
14. Choice of Sample Position
- After mounting a sample, find an obvious position on the sample, e.g. a corner, and note the position of the cryostat translations stages, by adding in the components as indicated by the blue dashed line in Figure 10.
- Move to another obvious position, e.g. along one of the edges, and note its position.
NOTE: Sample position is important when studying ensembles of filled carbon nanotubes. It is likely that the sample will be inhomogeneous. A good position on the sample has the following characteristics. - Find a location that is visually obvious when viewed through the microscope with detail on micron scale allowing you to return reliably to its position.
- Measure Raman signal from sample and adjust XY position slightly to check homogeneity of area on the few micron scale. If signal varies significantly choose a different spot.
- Capture an image of the spot and note the relative coordinates from sample corner to be able to return to sample.
15. Post Data Processing
- Import the spectra into data handling software.
- Normalize each spectrum with respect to laser power and exposure time to obtain counts per sec per mW.
- Correct for the spectrometer efficiency.
NOTE: Some spectrometer software does this automatically but, if not, then follow the following procedure. Steps 15.3.1-15.3.2 can be done once for any particular spectrometer and then used as necessary later.- Place a calibrated white light source at the position of the sample and measure its spectrum with all standard settings of the spectrometer.
- Divide the white light spectrum with the calibration curve for the white light source to obtain throughput of the spectrometer. Store the throughput files for later use.
- Divide the measured Raman spectrum by the throughput of the spectrum under identical conditions.
- Plot the Raman spectra using appropriate data handling software.
- Check spectra for evidence of the laser jumping between energies during the spectrum. This will produce broader peaks or a doubling up of the peaks in a spectrum.
- Check the Raman shift calibration by checking a known Raman feature, e.g. silicon LO (Longitudinal Optical) phonon, or checking for sudden changes in the shift of all of the Raman features in related spectra by the same amount.
- Fit Raman features using standard line shapes and a non-linear curving fitting program to obtain amplitude, center shift and width for all features.
- Plot the fit parameters versus laser energy or temperature as required.
Representative results for a series HRTEM images and simulations on a sample of HgTe@SWCNTs are displayed in Figure 1. The images throughout Figure 1A–F, depict low dimensional, confined HgTe extreme nanowires, with a diameter of ~1 nm, whose microstructure corresponds to the form discussed in ref 14. Representative images of bundles and discrete tubes are presented in Figure 1D. As per the protocol, a trial model is generated and simulated throughout various tilt angles and beam orientations, representative results of this is indicated in Figure 1A, B, C. These image simulations can be cross-correlated to real experimental results (Figure 1D, F) and can be seen to be a good match with simulations.
The main aim of the experiments described in this paper is to measure Raman spectra from extreme nanowires like those presented in Figure 2. The spectra presented in Figure 2 were measured using a sample of HgTe extreme nanowires taken from the same growth batch as the nanowires present in Figure 1. The sample was prepared for Raman using the method set out in sections 6 and 7 of the protocol. The spectra presented in Figure 2 show a large number of peaks most of which can be attributed to extreme nanowire vibrational excitations and multi-phonon Raman involving overtones and combinations of these vibrational excitations. The fundamental vibrational modes, A (45 cm-1), B (52 cm-1), C (94 cm-1) and D (115 cm-1), and some of their combinations and overtones are labeled on the spectra visible up to at least 6th order. The detailed attribution and interpretation of the HgTe Raman spectra are set out in reference14. It should be noted that strong multiple phonon Raman is a common feature of II-IV materials, such as HgTe, and not necessarily a feature of all extreme nanowire samples. In addition to the nanowire features the Raman spectra also contain one carbon nanotube Raman feature; predominantly due to a Radial Breathing Mode observed at 168 cm-1 whose resonant energy of 1.67 eV14 is clearly different from the resonance energies of the filling Raman features (Figure 4). The host tube Raman features can be clearly identified from Raman spectra of the pure nanotubes used for filling. A resonance Raman investigation of the unfilled tubes with a broader range of excitation energies is shown in the supplementary material along with an initial attribution of the 5 RBMs identified in this data.
The data presented in Figure 2 demonstrates the strong excitation laser energy dependence that is common in 1D system. This energy dependence is one of the key indicators that any Raman features observed are due to extreme nanowires rather than other forms of the parent material, or its thermal decomposition products, which remain in the sample after cleaning. Another key indicator is that the observed features are quite different from those of bulk HgTe30 which are dominated by a Longitudinal Optical (LO) phonon mode at 137 cm-1. There is significant evidence in the literature that Raman spectra of nanoparticles of HgTe with diameters down to 3 nm are dominated by bulk LO phonon derived vibrational modes and the same is true of HgTe quantum wells with dimensions down to 2 nm. The final key indicator that specific Raman features are associated with nanowires rather than nanoparticles or lumps of the parent material is a characteristic polarization dependence like that shown in Figure 3. As discussed in more detail in reference14 the Raman scattering from an ensemble of randomly oriented 1D systems is preferentially polarized in the same direction as the exciting laser light with a contrast ratio of 3:1 and thus shows the characteristic figure of eight shape present in the optimum results shown in Figure 3. It is important to test that preferred emission direction rotates with the excitation polarization, as shown in Figure 3, as polarized Raman due to other mechanisms is not uncommon. It is quite possible to observe a contrast ratio lower than 3:1 for thick layers of nanowires, as also shown in Figure 3, and this can be attributed to scattering of light within the layer.
Another possible explanation of Raman peaks in filled tube samples that are not present in unfilled tubes and not due to residual filling is that filling or residual material leads to modifications of the SWCNT Raman spectrum. For instance samples of SWCNTs which have had metal evaporated onto them exhibit "Squash" vibrational modes.31,32 However in the case of the HgTe filled samples we observe the opposite polarization dependence (Figure 3) to that observed for Squash modes.31 In addition the fact that high harmonics of the fundamental modes are observed in HgTe spectra and not for the Squash mode spectra allows us to rule out a Squash mode explanation for the HgTe Raman features.
The excitation photon energy dependence of the intensity of the B Raman feature taken from a full Resonance Raman experiment using the protocol set out in this paper is presented in Figure 4. Also presented is the same result from an experiment performed before the protocol was fully developed. With the protocol it is possible to get a variation in repeated, independent measurements of a single point on the resonance profile of approximately 8% as shown in Figure 5. The main parts of the alignment of the system that need to be controlled in order to get good quality spectra are alignment of the laser beam into the microscope objective and then focusing of the beam onto the sample. The importance of beam alignment is illustrated in Figure 6a, e. In this figure Raman spectra are shown (Figure 6a, blue trace) with the beam correctly aligned onto the two beam steering cameras (c and e) and sub-optimal spectra (Figure 6a, green trace) with the beam deliberately misaligned. A line through the vertical and horizontal center point of each of the frame b, e de Figure 6 shows there is small horizontal drift in the laser alignment as illustrated when 6b and 6d are compared. Comparing the green and blue traces in 6a, it is clear that a small misalignment can lead to significant variation (>50% loss) of Raman signal hitting the CCD.
The importance, and relevance, of using the reflected beam intensity to ensure the objective is correctly focused on the sample is illustrated in Figure 7. This figure presents the Raman intensity and reflected light signal as a function of the distance between the objective and the sample. To be within 10% of the peak Raman, the precision of the Z-position (the distance between the objective and sample) needs to be better than 20 µm, which is considerably larger than the distance between the peak positions of power and Raman as presented in Figure 7.
As discussed in the protocol it is important that the effect of laser excitation intensity on the Raman spectra are taken into account and that the experiment be in the regime in which the Raman scattering is proportional to the excitation intensity when measuring resonance profiles. Representative measurements of the excitation intensity dependence of the Raman scattering intensity of HgTe extreme nanowires, measured as per section 9 of the protocol, is shown in Figure 8. As presented in Figure 8 the Raman intensity initially increases linearly with excitation intensity up to an intensity of 1.5 x 104 before starting to show non-linear behavior with a tendency for the signal to saturate. The precise excitation intensity behavior of different samples will differ and so must be measured and taken into account. From Figure 8 the Raman intensity is clearly within the non-linear regime for excitation intensities greater than ~0.2 mW/mm2. Also shown is a linear fit to the data at low excitation intensities demonstrating that at sufficiently low enough excitation intensities the Raman intensity is proportional to excitation intensity (up to ~0.1 mW/mm2). It is important to reiterate this data is unique for this particular sample position at a particular temperature (4 K) and the experiment must be repeated as per the protocol steps when a different sample/temperature is investigated. As a general rule of thumb, it is ideal to use about 80% of the maximum power in the linear regime.
Once high quality Resonance energy dependence profiles have been measured these can then be analyzed to obtain a range of information. The theory underlying Raman processes is well understood and time dependent perturbation theory17, often calculated using a Feynman diagram approach21,33, can be used to predict resonance profiles and even absolute intensities. In the limit that the optical transitions are discrete and well separated in energy the theory predicts that the Raman intensity for single phonon scattering follows a Lorentzian lineshape centered at the optical transition multiplied by one centered one phonon energy above for Stokes scattering or one phonon energy below for Anti-Stokes Raman scattering. If the energy of the phonon is small compared to the resonance linewidth, as is the case for HgTe nanowires, this will lead to the Resonance having a Lorentzian squared lineshape. However in 1D systems it is likely that the features in the optical spectrum will be associated with van Hove singularities consisting of a continuum of states. In addition there is likely to be inhomogeneity within the sample further broadening the transition. If either or both of these are true then the density of states for the optical transitions will alter and can dominate the lineshape. The situation is made more complex because Raman scattering is a coherent process and so interference effects involving different scattering sequences and different intermediate states will alter the resonance profile34. For the same reason any variation of coherent lifetime amongst the intermediate states can also affect the lineshape35. The possibility of the involvement of elastic scattering from defects and double resonance effects, particularly in higher order Raman scattering, further complicates the situation21,35. It is often therefore not possible to a priori predict the expected Raman resonance profile. However resonance Raman scattering has been used to extract a great deal of information about different materials systems including the energy of features in the optical spectrum, the nature of the state's responsible for those features and the nature and quantitative strength of electron-phonon interactions17. In order to better quantify the energy and energetic width of the optical features within the Resonance profile it is often helpful to fit them using one of the standard optical lineshapes. In the case of the HgTe nanowires we tried Lorentzian, Lorentzian squared and Gaussian lineshapes and found the Gaussian lineshapes to be the best fit (Figure 4). To be clear this is a phenomenological fit and the use of the Gaussian lineshape cannot be interpreted in terms of the nature of the broadening of the optical feature which is causing the resonance. From these fits we can determine the energy of the optical feature responsible for the resonance to be 1.76 eV. A more detailed analysis of the resonance behavior of HgTe extreme nanowires will be published separately.
The measurement of the temperature dependence of Raman spectra enables additional physics to be probed. In particular the shift of the vibrational energies and the width of vibrational peaks allows for anharmonic effects, leading to lattice dilation and fundamental limits on the lifetime of phonons to be investigated. Measurements of Resonance profiles as a function of temperature will allow the temperature dependence of optical energies to be determined. Some representative results illustrating possible temperature related effects are presented in Figure 9. It can be seen from Figure 9 (a and b) that as temperature increases the spectral width broadens and the center shift of the mode softens, which is in-line with theoretical predictions. The most striking is window c, indicating a dramatic drop-off in intensity of the B mode as a function of temperature. This effect, which will be discussed in more detail in a separate publication, is predominantly due to a decrease in the coherent lifetime of the optical states responsible for the resonance with increasing temperature and is clear evidence that Raman scattering can provide information far beyond that possible with absorption measurements.
In order for the intensity of Raman scattering at different temperatures to be directly compared, drift of the lateral position of the sample needs to be corrected for. The inclusion of a light source and camera to allow the sample to be viewed through the microscope objective allows for the repositioning of the sample. If a "good" sample position is chosen as per step 14 of the protocol then it is possible to reposition the sample and achieve a reproducibility of the intensity of the Raman peak of better than 8% as shown in Figure 5.
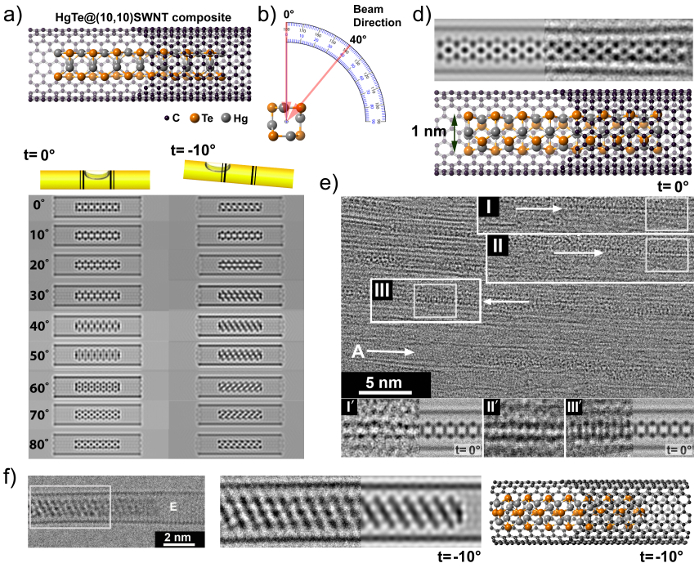
Figure 1: HRTEM of extreme nanowires with comparison to simulation results. Structure models, HRTEM simulation protocol and experimental images of ~1 nm thick HgTe nanowires embedded in ~1.4 nm diameter SWNTs. A typical partial cutaway model (a) of a 3 nm long fragment of HgTe embedded in a (10,10) SWNT. Electron beam directions (b) represent different projections for a series of orientations of the HgTe@(10,10)SWNT composite (c, LH simulations) and tilt (d, RH simulation). HRTEM images (e, top right) can be matched against the table (c) and matched with the experimental image (d top left and right). HRTEM image obtained from a thin SWNT bundle (e), used to observe embedded ~1 nm HgTe nanowires (I, II and III) and correlated with simulations in the tableau (i.e. insets I', II', and III'). Some fragments are tilted (f, left) by angle t, modeled by simulation (f, middle) that corresponds to the cutaway model as in f right. Please click here to view a larger version of this figure.
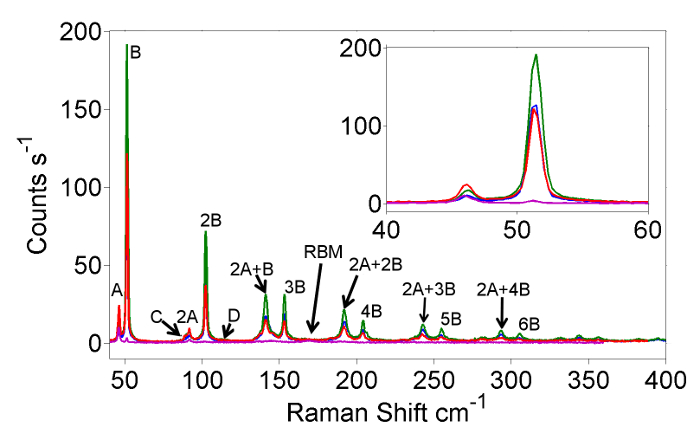
Figure 2: Raman spectra of extreme Mercury Telluride embedded within nanotubes. Representative Raman spectra of HgTe extreme nanowires in SWCNTs acquired at 4 K with multiple excitation photon energies. The various traces correspond to excitation energies of 1.78, 1.77, 1.75 and 1.71 eV for the blue, green red and purple lines respectively. Please click here to view a larger version of this figure.
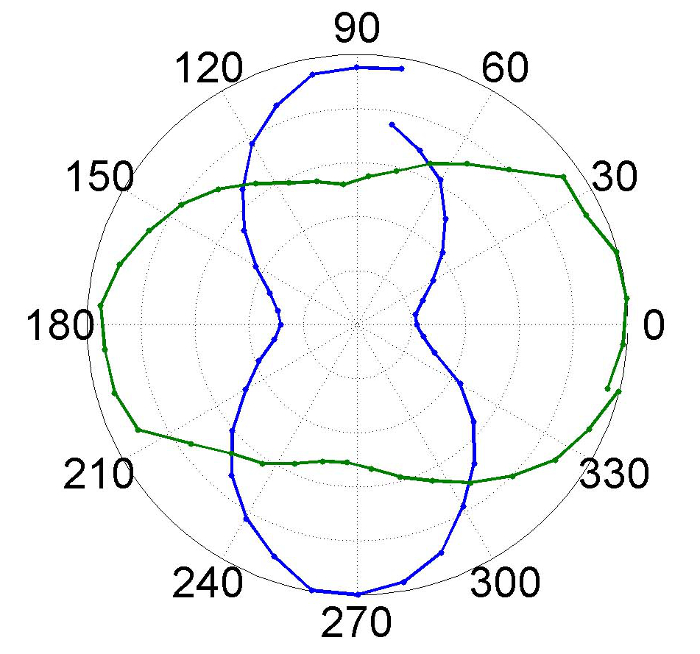
Figure 3: Fitted intensity of B peak as a function of analyzer angle. Polar plot of fitted intensity of the B peak at 1.77 eV and 4 K as a function of analyzer angle at vertical (blue) and horizontal (green) incident polarization. Please click here to view a larger version of this figure.
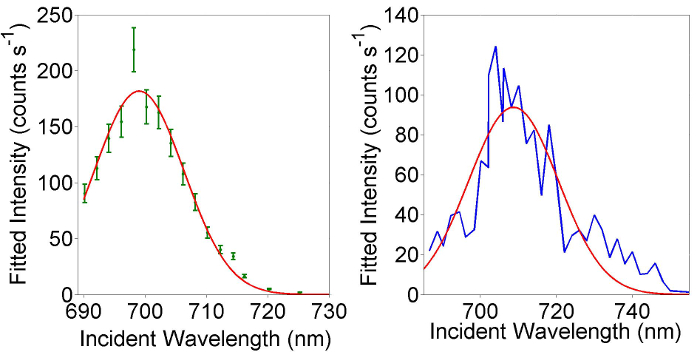
Figure 4: Resonance effects observed in B mode of HgTe@SWCNTs. Resonance profile of B (52 cm-1) mode as a function of laser wavelength for both (a) the case where the protocol detailed is adhered to and the case (b) taken before the protocol was developed. The Gaussian line widths are centered around 1.77 ± 1 meV and 1.74 ± 3 meV for a and b respectively. The errors were determined by the 95% confidence bounds of the fitting routine. Please click here to view a larger version of this figure.
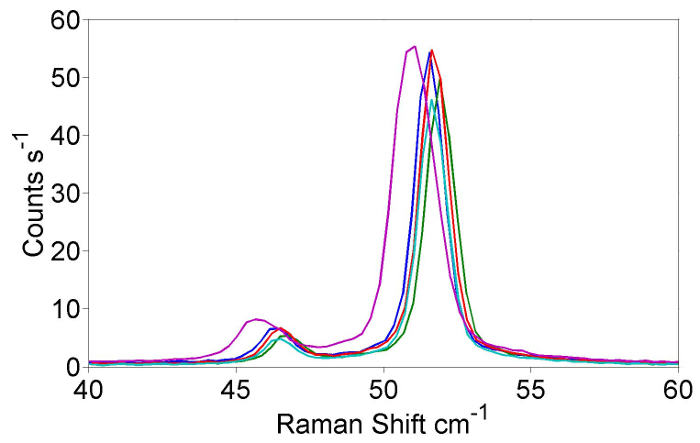
Figure 5: Independent repeats of Raman spectra at 702 nm incident wavelength throughout RRS experiment. A series of Raman spectra, taken under identical conditions throughout the experiment. The spectra show the A and B mode measured with a 702 nm laser line at 4 K during a resonance Raman experiment. Please click here to view a larger version of this figure.
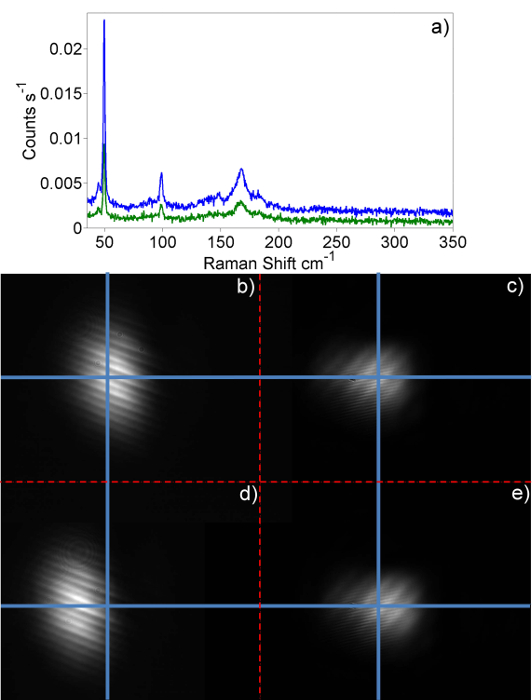
Figure 6: Raman spectra of HgTe@SWCNTs taken when system is optimized and deliberately de-tuned. Raman spectra acquired at room temperature when the system is well aligned (blue trace) and deliberately misaligned (green trace). Frames (b, d) show laser image on camera (C2) and (c, e) show laser spot on camera (C1). The well-aligned spectra correspond to the images from b and c whilst the deliberately misaligned spectra are shown through d and e. Please click here to view a larger version of this figure.
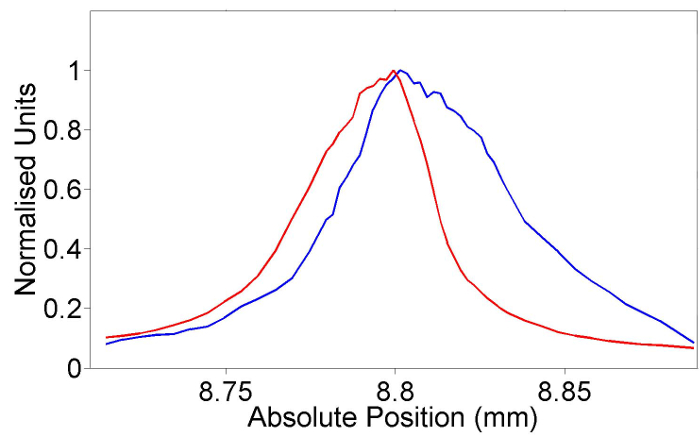
Figure 7: Reflected power and corresponding Raman peak intensity of Si peak as a function of sample focal-position. Plot of the normalized reflected power (red) measured on power meter (PM2) and the normalized intensity of Raman intensity (blue) as a function of distance between sample and objective. Please click here to view a larger version of this figure.
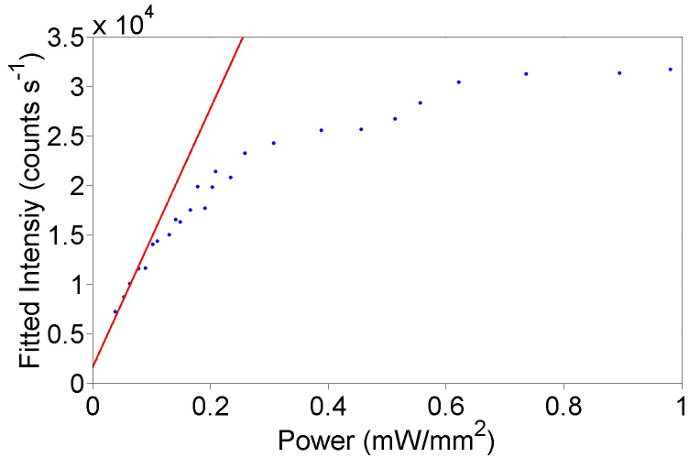
Figure 8: Plot of the intensity of the B Raman mode at 4 K and 702 nm quantified with Lorentzian fitting as a function of excitation intensity. The fitted intensity of the B mode as a function of incident power, where a fit is applied to determine the linear regime. Please click here to view a larger version of this figure.
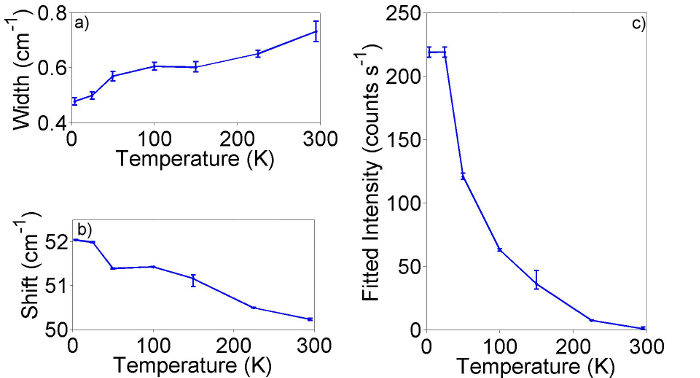
Figure 9: Temperature dependence of B mode in HgTe@SWCNTs at fixed (1.77 eV) excitation energy. Raman spectra acquired at constant excitation energy (1.77 eV) as a function of temperature. Windows a-c shows the spectral width, center shift and fitted intensity of the B modes respectively. Error bars shown are the 95% confidence bounds from the fitting routine. Please click here to view a larger version of this figure.
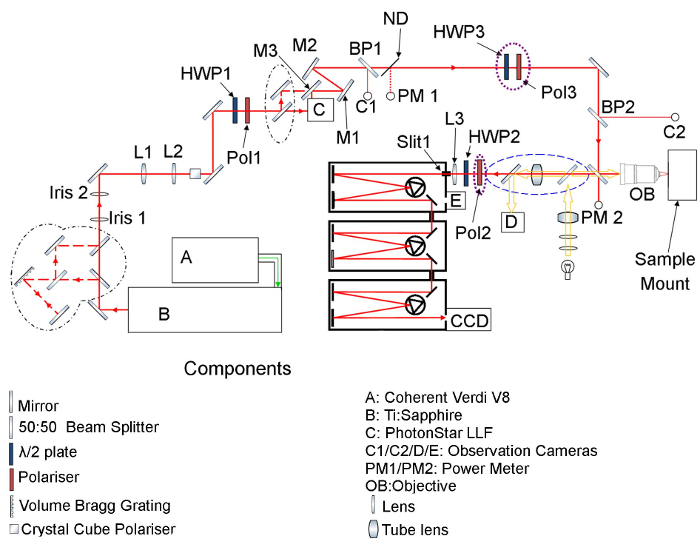
Figure 10: Schematic of optical setup employed for resonance Raman Spectroscopy experiments. Figure displays optical setup employed for all experiments discussed in protocol. Please click here to view a larger version of this figure.
