Time-lapse microscopy is a powerful technique to watch dynamic biological processes over a longer period of time. An often occurring problem during time-lapse imaging is a movement in x, y or z direction, called drift, which is induced by temperature variations and mechanical vibrations. The method presented here enables time-lapse recording of fluorescently labeled structures in zebrafish embryos or larvae on a dissecting microscope without environmental chamber and anti-vibration table.
For compensation of xy-drift, the specimen was embedded in an imaging chamber with an imprinted relocation grid (Figure 1). The location of a particular point of the grid related to the crosshairs of the software tool was used to return the specimen to the pre-adjusted position (Figure 2A). The presented results were obtained by using a zoom microscope with an integrated slider for optical sectioning via structured illumination (Figure 2B). For continuous focal correction, an autofocus strategy was established. In this strategy, the software autofocus is used to find the focus based on maximum contrast before every time point. This so defined focal plane is subsequently set as center plan for z-stack acquisition. In order to minimize phototoxicity in the sample, the transmitted light brightfield channel is used as reference channel as it allows sufficiently short exposure times during the autofocus step (Figure 2C).
The method was applied for comparative analysis of kidney development in control- and wt1a morphant embryos of a transgenic zebrafish line (Tg(wt1b:GFP)). During early nephrogenesis this line shows green fluorescence in the intermediate mesoderm, were the kidney progenitors arise from 9,10 and later in the forming tubules and nephron primordia (Figure 3).
Time-lapse image recording reveals that nephrogenesis in control morpholino injected embryos is unaltered in comparison to uninjected ones (results not shown) and follows the described steps of early zebrafish kidney development 3. At 20 hpf the developing pronephric tubules are visible and at their anterior tips, spherical accumulations of cells, representing the forming nephron primordia can be detected. During the next hours, tubules and nephron primordia grow and later on the primordia start to fuse at the midline (Figure 4, video 2). In contrast, nephrogenesis is severely disrupted in wt1a morphant embryos. Although GFP-positive tubular structures are visible at 20 hpf, they appear to be more diffuse and less developed. Furthermore, no proper nephron primordia have been formed. The most striking difference to the control embryos at this time point, however, is the appearance of a large number of fluorescent cells outside the developing pronephros (Figure 4, video 2). Subsequently, these cells leave the pronephric field and migrate ventrally (video 2).
Kidney development in control embryos and wt1a morphants of the wt1b transgenic line have been previously compared by taking images at different fixed time points 9,10. In contrast to this static method, time-lapse recording allows to follow dynamics of normal nephrogenesis and misrouting of kidney progenitor cells caused by wt1a depletion.

Figure 1: Schematic Illustration of an Embedded Embryo in a Commercially Available Chamber with Relocation Grids. The dish has four grids, each subdivided into a repeat distance of 50 µm, imprinted into a glass coverslip bottom. The embryo to be imaged is embedded upside down, with kidney structure in close proximity to an observation square but without overlaying with the grid. Please click here to view a larger version of this figure.
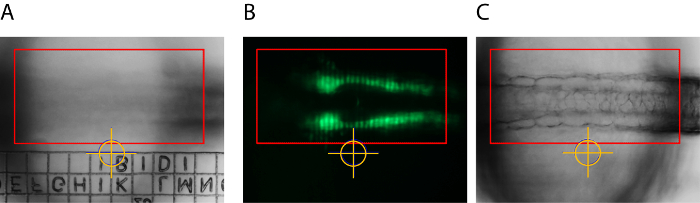
Figure 2: Adjustments for Compensation of Drift in Time-lapse Imaging and Grid Projection of Structured Illumination. A distinct position of the relocation grid was brought into the image center (marked by yellow crosshairs) and serves as a reference point for later xy-alignment in case of small shifts. The red rectangle represents the region of interest (ROI) used in the autofocus strategy (A). Optical sectioning was obtained by structured illumination. The image shows a grid structure projected in the focal plane after correct calibration (B). The ROI for the autofocus strategy (red rectangle) is set to cover distinctive embryonic structures high in contrast (here somites) that allow reliable autofocusing into the structure of interest (C). Please click here to view a larger version of this figure.
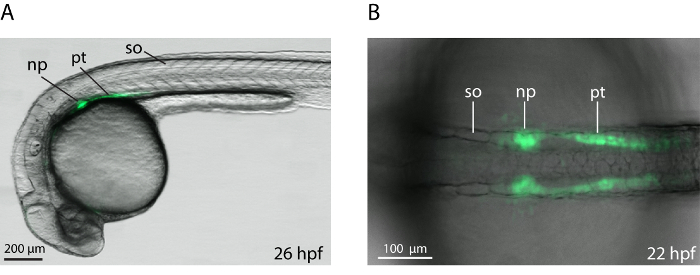
Figure 3: Transgenic wt1b:GFP Embryos with Green Fluorescence in the Developing Kidney. Overlays of dorsal (A) or lateral (B) transmission and fluorescence images are shown with anterior to the left. np, nephron primordium; pt, pronephric tubule; so, somite. Please click here to view a larger version of this figure.
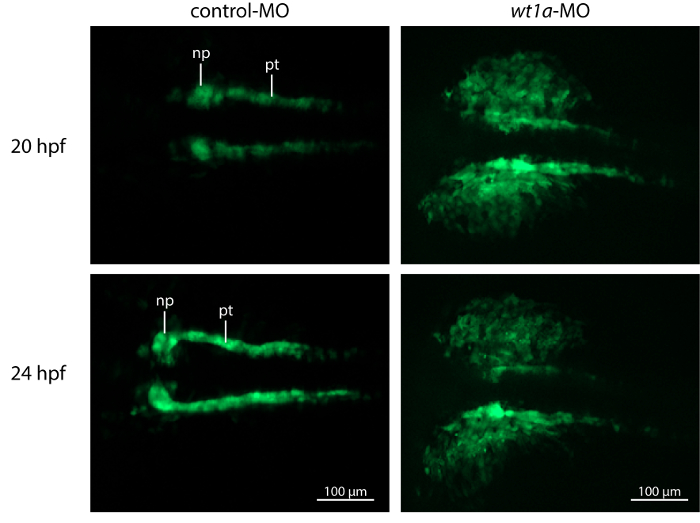
Figure 4: Knockdown of wt1a Disrupts Embryonic Kidney Development. Representative, extended depth of focus images from time-lapse recordings. In control morpholino injected embryos, kidney development shows normal progress with growing tubules and nephron primordia which start to fuse at the midline. In contrast, wt1a morphants fail to form proper nephron primordia and a massive amount of GFP positive cells are outside of the pronephric field. (np, nephron primordium; pt, pronephric tubule). Please click here to view a larger version of this figure.
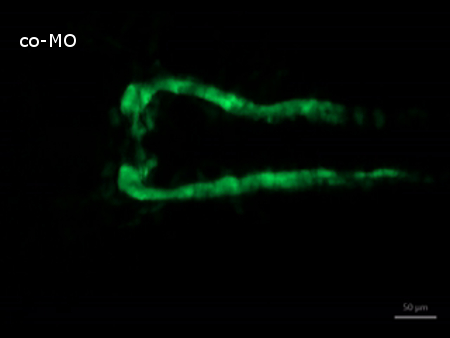
Supplemental Video 1. Time-lapse recording of normal kidney development in control morpholino injected embryos. (Right click to download). The video shows how tubules grow and nephron primordia begin to fuse. Starting at 20 hpf, images were taken in 30 min intervals over a period of 5 hr.
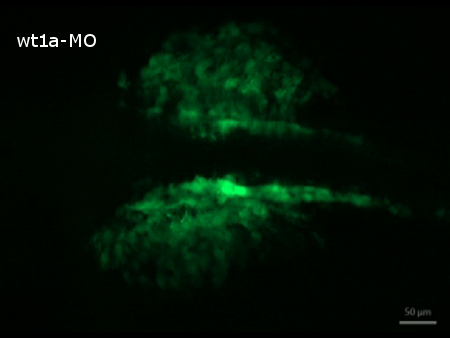
Supplemental Video 2. Time-lapse recording of disturbed nephrogenesis in wt1a morphant embryos. (Right click to download). The video shows the migration of GFP-positive cells out of the pronephric region. Starting at 20 hpf, images were taken in 30 min intervals over a period of 5 hr.
