This protocol is a series of established methods linked together to allow for the quantification of sRNAs on highly pure HDL by high-throughput sequencing or real-time PCR (Figure 1). To demonstrate the feasibility and impact of this protocol, HDL was purified from human plasma by the tandem DGUC and FPLC method. Collected FPLC fractions corresponding to HDL (by cholesterol distribution) were concentrated and total RNA was isolated from 1 mg of HDL (total protein). sRNA libraries were generated with HDL collected from n = 4 healthy human subjects. All human blood/plasma samples were collected under active Institutional Review Board protocol (# 070416), and human subjects were enrolled with informed consent as approved by Vanderbilt University School of Medicine. Prepared sRNA libraries were sequenced at Vanderbilt Technologies for Advanced Genomics (VANTAGE) DNA sequencing center and data analyses were performed using in-house pipelines. Data visualizations were created by graphing software.
The critical need to perform a second method (FPLC) for HDL isolation after DGUC is due to possible co-purification of EVs. EVs are a generalized class of membrane-derived vesicles that originate from multivesicular bodies (exosomes), plasma membrane budding (microvesicles), and other processes, including apoptotic bodies10. EVs, namely exosomes, have been reported to have a similar density as small HDL (Figure 2A), and thus, could be present in the HDL density fraction (1.063 – 1.21 g/mL) after DGUC11,12. To remove EVs and other lipoproteins (e.g., LDL), from DGUC-HDL, FPLC was used to separate HDL from other vesicles and particles by size; HDL (7 – 12 nm in diameter) and EVs (40 – 220 nm in diameter) are easily fractionated due to their size differences (Figure 2A,B). HDL fractions were readily apparent due to the distribution of cholesterol, and DGUC-HDL FPLC fractions were pooled and concentrated using 10 kDa cut-off centrifugation filters. Concentrated HDL were collected and used for downstream RNA analysis (Figure 2B). To illustrate this point, plasma pre-DGUC was fractionated using FPLC and the distribution of phospholipids (phosphatidylcholine, PC); total cholesterol (TC), and protein levels were quantified and plotted across fractions. PC, TC, and proteins levels were quantified using three colorimetric kits. All three parameters clearly defined the HDL fractions (19 – 24) and VLDL/LDL fractions (14 – 18). In FPLC-fractionated plasma, protein and lipid signals were minimally detected or not detected at all in fractions corresponding to vesicle sizes greater than VLDL (left of orange box) (Figure 3A). To demonstrate DGUC separation of HDL, DGUC-HDL was also fractionated by FPLC and PC, TC, and proteins levels were quantified (Figure 3B). Plotting these values illustrates the co-fractionation of a small amount of LDL using DGUC and reinforces the need to use the tandem DGUC-FPLC approach to isolate highly pure HDL. Although EVs are predicted to co-fractionate with HDL during DGUC, there was little to none found in the lipid and protein in fractions corresponding to vesicle sizes greater than LDL (Figure 3B). The EV lipid and protein signature in the predicted size range is also minimally detectable or absent from DGUC-VLDL and DGUC-LDL when fractionated by FPLC (data not shown). For RNA analysis, total RNA was isolated from 1 mg of tandem purified HDL for sRNA library generation. Briefly, adapters were ligated to the 3' and then 5' terminal ends of sRNAs, including miRNAs and tDRs (Figure 4A). Ligated products were reverse transcribed (RT) and PCR amplified using PCR primers harboring specific indexes designed for multiplexing samples during downstream sequencing reactions (Figure 4A). Each adapter is 61 nts in length; therefore, a ligated miRNA (approximately 22 nts in length) would produce a product of 144 nts in length. Many non-miRNA sRNAs are slightly longer than miRNAs (e.g., 30 – 35 nts in length). As such, our targeted length of product was 156 bps in length. Amplified products were size-selected using an automatic DNA size selector and all cDNA products 135 – 177 bps in length were collected (Figure 4A). The qualities of size-selected libraries were assessed by high-sensitivity DNA chips using the bioanalyzer (Figure 4B). sRNA cDNA libraries of this size appeared slightly larger in size due to possible "bubbling" effects during the analysis. For multiplexed sequencing, equimolar concentrations of cDNA libraries were pooled, cleaned, concentrated, and reanalyzed using the bioanalyzer (Figure 4B). The multiplexed sample was then sequenced using a single read 50 bp protocol. In-house data analysis pipelines were used to demultiplex samples based on indexes, trim adapters, run quality control metrics, align to the human genome and count annotated sRNAs. The distribution of normalized reads (Reads Per Million, RPM), > 16 nts in length, illustrate the enrichment of sRNAs 20 – 22 nts in length and 34 – 35 nts in length on HDL (Figure 4C).
Alignment and counting analysis of the four HDL-sRNA libraries found that 38% of reads aligning to annotated regions of the human genome were miRNAs (Figure 5A). Equally abundant (37%) were sRNAs-derived from tRNAs (tDRs). sRNAs-derived from miscellaneous RNAs (e.g., Y RNAs), rRNAs, smRNAs, snoRNAs, and long non-coding RNAs (lincRNAs) were also present on HDL (Figure 5A). After normalization to the total number of reads for each class, the top 50 most abundant miRNAs (Reads Per Mapped miRNA, RPMM, Figure 5B), tDRs (Reads Per Mapped tDRs, RPMT, Figure 5C), and other non-miRNA non-tDR sRNAs (Reads Per Mapped sRNA, RPMS, Figure 5D) were organized by heatmaps. These data illustrate the similarities (e.g., HDL-miRNAs) and discrepancies (e.g., tDRs) of the most abundant HDL-sRNAs across healthy subjects (Figure 5B-D). These results demonstrate the power of this strategy and protocol to provide remarkable insights into the transport of sRNAs by HDL. Nevertheless, sRNA sequencing should not be relied on for absolute quantitation. sRNAs of interest should be validated by real-time PCR. Likewise, many investigators may only require the quantification of individual miRNAs on HDL. As such, total HDL RNA was isolated from each subject for relative quantification of HDL-miRNAs using real-time PCR. Five of the more abundant miRNAs detected on HDL by sRNAseq were used for real-time PCR assays to determine their levels on each HDL sample (Figure 5E). The relative quantitative values were calculated using an arbitrary housekeeping Ct of 32 (RQV= 2–ΔCtArb) and normalized to 1,000 µg of HDL protein input into the RNA isolation13. Results from the real-time PCR study demonstrate that this protocol is also valuable in detecting HDL-miRNAs and quantifying differences across individuals. Collectively, the presented schematics define the critical details of the methods, and the results from both high-throughput sequencing and real-time PCR approaches represent the deliverables of this protocol.
Figure 1. Representative Flow-chart of the Protocol. Pure HDL is isolated from density gradient ultracentrifugation and fast protein liquid chromatography. Total HDL RNA can then be utilized for small RNA (sRNA) sequencing. Please click here to view a larger version of this figure.
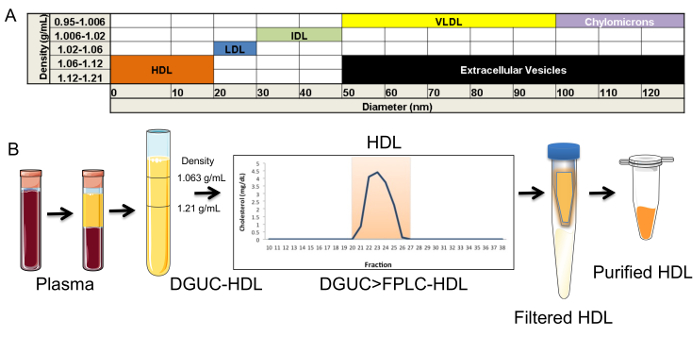
Figure 2. Isolation of HDL. (A) Chart of the density and diameter of lipoproteins and extracellular vesicles (EV). (B) Schematic of sequential tandem HDL isolation, including plasma separation, density-gradient ultracentrifugation (DGUC), fast-protein liquid chromatography (FPLC), filtration, and concentration. Please click here to view a larger version of this figure.
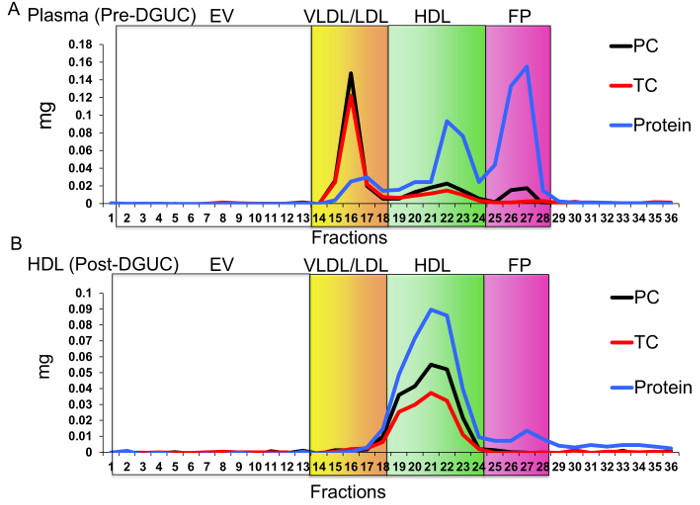
Figure 3. Fast-protein Liquid Chromatography (FPLC) Separation of Lipoproteins. EV, extracellular vesicles; VLDL, very low-density lipoproteins; LDL, low-density lipoproteins; HDL, high-density lipoproteins; FP, free protein; PC, phosphatidylcholine; and TC, total cholesterol. Red line, TC; Blue line, protein, and Black line, PC. Separation of lipoproteins from (A) human plasma prior to density-gradient ultracentrifugation (DGUC) and (B) HDL fraction after DGUC. Please click here to view a larger version of this figure.
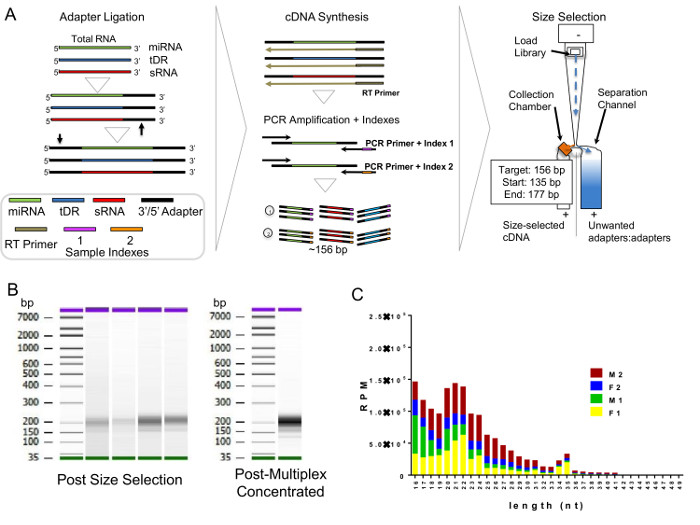
Figure 4. Generation of Small RNA Libraries. (A) Schematic of small RNA (sRNA) library construction and size-selection. miRNA, microRNA; tDR, tRNA-derived sRNAs; sRNA, non-miRNA non-tDR sRNAs; RT, reverse transcription; bp, base pair; and cDNA, cloned DNA. (B) Analysis of HDL-sRNA libraries before and after multiplexing using high-sensitivity DNA chips and bioanalyzer. (C) Length distribution of HDL-sRNA reads after sequencing. RPM, Reads Per Million; M, male subjects; F, female subjects; and nt, nucleotide. N=4. Please click here to view a larger version of this figure.
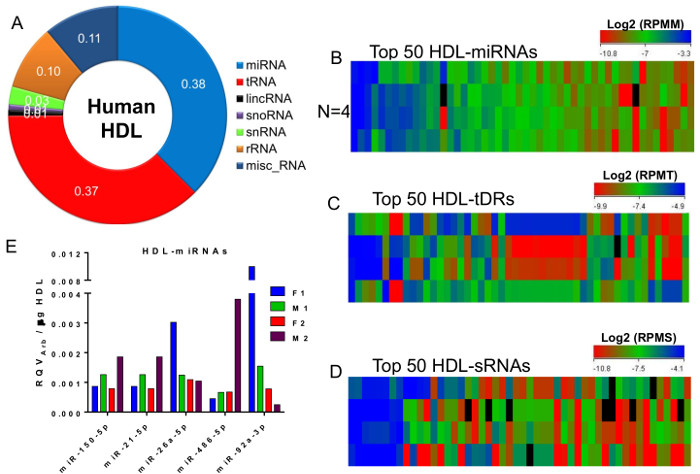
Figure 5. Diversity of HDL-sRNAs. (A) Distribution of HDL-sRNAs across sRNA classes. Fraction of reads aligned to annotated sRNAs in the human genome. (B-D) Heatmaps N = 4 HDL-sRNA samples. (B) Top 50 most abundant HDL-microRNAs (miRNA). Log2 Reads Per Mapped miRNA, RPMM. (C) Top 50 most abundant HDL-tRNA-derived sRNAs (tDRs). Log2 Reads Per Mapped tDR, RPMT. (D) Top 50 most abundant HDL-other non-miRNA and non-tDR sRNAs (sRNA). Log2 Reads Per Mapped sRNA, RPMS. (E) Real-time PCR quantification of HDL-miRNAs. M, male subjects; F, female subjects. Relative Quantitative Value determined by arbitrary housekeeping Ct = 32, normalized to 1,000 µg of HDL protein input13. N = 4. Please click here to view a larger version of this figure.
| 1. Anti-oxidants for Lipoprotein separation | Make stock solution at 100x |
| EDTA | Ethylenediaminetetraacetic acid (0.5 M stock) |
| AZT | 3-Amino-1,2,4-triazole: 42 mg/mL H2O |
| BHT | Butylated Hydroxytoluene: 25 mg/100 µL EtOH |
| TROLOX | (+)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid: 41.7 mg/500 µL EtOH |
| 2. Overlay Solutions | |
| Solution #1 | 1.019 g/mL in H2O |
| Solution #2 | 1.025 g/mL KBr/H2O |
| Solution #3 | 1.063 g/mL KBr/H2O |
| Solution #4 | 1.080 g/mL KBr/H2O |
| Solution #5 | 1.255 g/mL KBr/H2O |
| 3. FPLC Buffer | |
| 1 L Recipe | 50 mL 3M NaCl; 20 mL 0.5 Tris-HCl, pH 7.6; 2 mL of 10% Sodium Azide; 930 mL H2O |
Table 1. Special Reagents.
| Stage | Temp (°C) | Time | Cycles |
| A | 98 | 30 s | 1 |
| B | 98 | 10 s | 25 |
| 60 | 30 s | ||
| 72 | 15 s | ||
| C | 72 | 10 min | 1 |
Table 2. Library Generation Amplification Steps.
| Primer | Sequence |
| Primer 1.1 | AATGATACGGCGACCACCGAGAT |
| Primer 2.1 | CAAGCAGAAGACGGCATACGA |
Table 3. PCR Primers.
| Stage | Temp (°C) | Time | Cycles |
| A | 95 | 10 min | 1 |
| B | 95 | 10 s | 40 |
| 60 | 30 s |
Table 4. Library Quantification Amplification Steps.
