An Optimized Protocol for Electrophoretic Mobility Shift Assay Using Infrared Fluorescent Dye-labeled Oligonucleotides
Summary
We describe here an optimized protocol of fluorescent Electrophoretic Mobility Shift Assays (fEMSA) using purified SOX-2 proteins together with infrared fluorescent dye-labeled DNA probes as a case study to tackle an important biological question.
Abstract
Electrophoretic Mobility Shift Assays (EMSA) are an instrumental tool to characterize the interactions between proteins and their target DNA sequences. Radioactivity has been the predominant method of DNA labeling in EMSAs. However, recent advances in fluorescent dyes and scanning methods have prompted the use of fluorescent tagging of DNA as an alternative to radioactivity for the advantages of easy handling, saving time, reducing cost, and improving safety. We have recently used fluorescent EMSA (fEMSA) to successfully address an important biological question. Our fEMSA analysis provides mechanistic insight into the effect of a missense mutation, G73E, in the highly conserved HMG transcription factor SOX-2 on olfactory neuron type diversification. We found that mutant SOX-2G73E protein alters specific DNA binding activity, thereby causing olfactory neuron identity transformation. Here, we present an optimized and cost-effective step-by-step protocol for fEMSA using infrared fluorescent dye-labeled oligonucleotides containing the LIM-4/SOX-2 adjacent target sites and purified SOX-2 proteins (WT and mutant SOX-2G73E proteins) as a biological example.
Introduction
EMSAs are used to analyze interactions between DNA and proteins by using native PolyAcrylamide Gel Electrophoresis (PAGE) to resolve a mixture of a protein of interest and a labeled DNA probe containing potential target sites of the protein1. A DNA probe bound with protein will migrate slower compared with a free DNA probe, and is therefore retarded in its migration through a polyacrylamide matrix. Radiolabeling of DNA by 32P has been the predominant method for detection in EMSAs. Although the application of radiolabeling in biochemical research has been beneficial, methods of alternative DNA labeling with comparable sensitivity are being employed due to the health and safety risks associated with handling radioactivity. These alternative methods include conjugation of DNA with biotin2 or digoxigenin (DIG)3 (both of which are then detected by chemiluminescent systems), SYBR green staining of the PAGE gels4, or direct detection of DNA-fluorescent dye conjugates by scanning the gel5,6.
The resolved gels of EMSAs using radioactively labeled DNA probes require postrun processing through autoradiography films or a phosphorimager system to detect radioactive signals1,7. Gels of EMSAs using biotin-2 or DIG-conjugated3 DNA probes must also be processed and transferred onto a suitable membrane and then detected by chemiluminescence6. SYBR green staining of the gel requires postrun gel staining and a fluorescent scanner4. Since postrun gel processing steps are required for EMSAs using these DNA labeling techniques, the resolved gel can be assayed only once. In contrast, DNA probes labeled with fluorophores can be directly detected in the gel inside the glass plates by a scanner. Therefore, the DNA-protein interactions can be monitored and assayed several times at different time points during the run, which significantly reduces time and cost. The DNA-fluorescent dye conjugates that have been used for EMSAs include Cy36,8, Cy56,8, fluorescein9, and infrared fluorescent dyes4-6.
Transcriptional regulation requires protein-DNA interactions of transcription factors and their target genes. Coordination of these interactions generates diverse cell types from a common progenitor during animal development. An unbiased forward genetic screen identified a missense mutation, G73E, in the highly conserved HMG DNA-binding domain of the Caenorhabditis elegans transcription factor SOX-2. The mutation results in a cell identity transformation of AWB olfactory neurons into AWC olfactory neurons at molecular, morphological, and functional levels5,10. SOX-2 differentially regulates the terminal differentiation of AWB and AWC neurons by interacting with context-dependent partner transcription factors and respective DNA target sites5,10. SOX-2 partners with LIM-4 (LHX) in terminal AWB neuronal differentiation, while SOX-2 partners with CEH-36 (OTX/OTD) in terminal AWC neuronal differentiation. Luciferase reporter assays show that SOX-2 and mutant SOX-2G73E proteins have cooperative interactions with transcriptional cofactors LIM-4 and CEH-36 to activate a promoter expressed in AWB and AWC neurons. However, SOX-2 and mutant SOX-2G73E displayed differential activation properties of the promoter. To investigate the molecular basis of differential DNA binding activities of SOX-2 and SOX-2G73E, fEMSAs were performed with these proteins and their potential target sites.
First, a bioinformatics approach was taken to identify the biologically relevant DNA binding sequences within the 1 kb promoter region used in the luciferase assay. Since multiple potential SOX-2 binding sites were present throughout the promoter, we focused on predicted SOX-2 binding sites adjacent to putative CEH-36 or LIM-4 binding sites and evolutionarily conserved between various nematode species. These sequences were deleted or mutated, and subsequently tested in vivo for their activity to express GFP reporter transgenes in AWB or AWC neurons. Through this approach, we identified potential LIM-4/SOX-2 and CEH-36/SOX-2 adjacent target sites that are specifically required for the expression of GFP in AWB and AWC neurons, respectively5. We investigated the potential differences in the DNA binding of SOX-2 and SOX-2G73E using fEMSA with the identified LIM-4/SOX-2 and CEH-36/SOX-2 adjacent target sites. Our EMSA analysis showed that SOX-2G73E did not bind the DNA probe containing the LIM-4/SOX-2 adjacent target sites (required for gene expression in AWB) as efficiently as WT SOX-2 did. However, SOX-2 and SOX-2G73E had no difference in binding the DNA probe containing the CEH-36/SOX-2 adjacent target site (required for gene expression in AWC)5,10. Our fEMSA analyses provide mechanistic insight into the nature of the SOX-2G73E mutation in affecting specific DNA binding activity for the AWB-to-AWC cell identity transformation phenotype. Here, we describe an optimized protocol of fEMSA using purified 6xHis-SOX-2 or 6xHis-SOX-2G73E together with infrared fluorescent dye-labeled DNA probes containing the LIM-4/SOX-2 adjacent target sites as a case study to tackle an important biological question.
Protocol
NOTE: EMSAs using fluorescently labeled DNA probes or other forms of labeled DNA share the same protocols for protein or cell extract preparation, protein-DNA binding reaction, and PAGE gel preparation and running (Figure 1A). The key differences are DNA labeling procedures, post-run gel processing steps, and detection methods.
1. Gel Preparation
- Prepare 5% native polyacrylamide gel containing 0.5x TBE (45 mM Tris-Borate, 1 mM EDTA) and 2.5% glycerol, using a mini protein gel system (8.3 cm width x 7.3 cm length x 0.75 mm thickness).
- To prepare 30 mL of gel solution for 4 gels, mix 5 mL of 30% Acrylamide/Bis (37.5:1), 3 mL of 5x TBE (0.45 M Tris-Borate, 10 mM EDTA), 1.5 mL of 50% Glycerol, 300 µL of 10% Ammonium Persulfate, 15 µL of TEMED, and 20.2 mL of ddH2O thoroughly.
NOTE: Acrylamide is harmful and toxic, handle with appropriate personal protective equipment.
- To prepare 30 mL of gel solution for 4 gels, mix 5 mL of 30% Acrylamide/Bis (37.5:1), 3 mL of 5x TBE (0.45 M Tris-Borate, 10 mM EDTA), 1.5 mL of 50% Glycerol, 300 µL of 10% Ammonium Persulfate, 15 µL of TEMED, and 20.2 mL of ddH2O thoroughly.
- Cast the gel solution immediately. After polymerization, wrap the gels in clear plastic wrap pre-wetted with 0.5x TBE and store them at 4 °C.
2. Preparation of Infrared Fluorescent Dye-labeled Probes
NOTE: Keep the infrared fluorescent dye-labeled oligonucleotides away from light as much as possible during preparation, storage, and experiments.
- Design and order oligonucleotides.
- Design long oligonucleotide for ~51-mers.
NOTE: The long oligonucleotide sequence of the LIM-4/SOX-2 probe is TATCATATCTATTCTATGATTAAATACCTATTCATTTCAAAATCTTCTCCC. Long oligonucleotides do not require PAGE or HPLC purification. - Design complementary short oligonucleotides for ~14-mers with infrared fluorescent dye modification at the 5' terminus (5'Dye) and a melting temperature above 37 °C.
NOTE: The short oligonucleotide sequence of the LIM-4/SOX-2 probe is 5'Dye-GGGAGAAGATTTTG. The minimum synthesis scale of 5'Dye-labeled short oligonucleotides is 100 nM. Therefore, the oligonucleotides require HPLC purification.
- Design long oligonucleotide for ~51-mers.
- Resuspend oligonucleotides in 1x Tris-EDTA buffer (TE: 10 mM Tris-HCl, pH 8.0; 1 mM EDTA) to final concentration of 100 µM.
- Anneal Short (5'Dye) and Long oligonucleotides.
- Mix 0.6 µL of 5'Dye-Short oligo (100 µM), 1.2 µL of Long oligo (100 µM), and 28.2 µL of sodium chloride-Tris-EDTA buffer (STE: 100 mM NaCl; 10 mM Tris-HCl, pH 8.0; 1 mM EDTA) in a 1.5 mL tube.
- Place the tube in boiling water for 5 min. Turn off the heat source and let the water with the annealed oligos cool O/N in the dark.
- Fill in the 5' overhangs of annealed 5'Dye-Short/Long oligos with Klenow DNA polymerase to make double stranded DNA probes.
- Prepare the reaction by mixing 30 µL of annealed short/long oligos with 5'Dye tag, 8.5 µL of 10x Klenow buffer, 1.7 µL of 10 mM dNTPs, 1.7 µL of Klenow (3'->5' exo-) (5 u/µL), and 43.1 µL of ddH2O in a 0.2 mL PCR tube.
- Incubate 60 min at 37 °C in a PCR machine.
- Add 3.4 µL 0.5 M EDTA to stop the reaction and heat inactivate at 75 °C for 20 min in a PCR machine.
- Dilute filled-in probes (0.7 µM) in STE for final concentration of 0.1 µM.
- Store oligos at -20 °C in the dark until ready to use. Oligos may be stored for up to a year in this condition.
NOTE: To generate DNA probes with mutant binding sites, mutant versions of long oligonucleotides are annealed with 5'Dye-labeled short oligonucleotides, followed by fill-in of 5' overhangs with Klenow. It has been shown that the probe with one strand labeled with an infrared fluorescent dye has only 30% intensity of the probe with both strands labeled with the dye. If signal intensity needs to be strengthened, long oligos of both strands are labeled with infrared fluorescent dyes at 5' termini and annealed to make infrared fluorescent dye-labeled probes.
3. Preparation of Unlabeled/Cold Probes (Competitors)
Note: Long oligonucleotides of complementary sequences (Long and LongR) were annealed to make unlabeled probes for competition analysis with infrared fluorescent dye-labeled probes.
- Mix 20 µL of 100 µM Long, 20 µL of 100 µM LongR oligonucleotides, and 60 µL of STE in a 1.5 mL tube.
- Place the tube in boiling water for 5 min, turn off the heat source and let the water with the annealed oligos cool overnight.
4. Binding Reaction and Electrophoresis
- Prepare 1 mL of 5x binding buffer by mixing 50 µL of 1 M Tris-HCl, pH 7.5; 10 µL of 5 M NaCl; 200 µL of 1 M KCl, 5 µL of 1 M MgCl2, 10 µL of 0.5 M EDTA, pH 8.0; 5 µL of 1 M DTT; 25 µL of 10 mg/mL BSA, and 695 µL of ddH2O.
NOTE: The 5x Binding Buffer can be aliquoted and stored at -20 °C. Another point to consider is that different transcription factors will have different modifications to the binding buffer. - Right before setting up binding reactions, pre-run the 5% native polyacrylamide gel in 0.5x TBE + 2.5% glycerol to remove all traces of ammonium persulfate at 80 V for 30 - 60 min, or until the current no longer varies with time.
- Set up the binding reaction in final volume of 20 µL.
- Mix 4 µL of 5x binding buffer, 80 – 200 ng purified protein A (in 50% glycerol, i.e., SOX-2), 1 µL of 0.1 µM Dye-conjugated probe (final concentration: 5 nM), and ddH2O.
- Optional: When cold probe competitors are required, add various concentrations (2x, 10x, 25x, 50x, 100x, etc.) of cold probes.
- Optional: When the interaction between proteins A and B (i.e., SOX-2 and LIM-4) is tested, add 80 – 200 ng purified protein B (in 50% glycerol, i.e., LIM-4).
- Optional: When nuclear extract preparation is used and specific binding of protein A is to be validated, add antibody specific to protein A (i.e., anti-6xHis, anti-FLAG, etc.).
- Incubate at R/T in dark for 15 min.
- Load all of the binding reaction onto the gel and run the gel at 10 V/cm to desired distance.
5. Imaging
NOTE: The gel was scanned directly in the glass plates with an advanced infrared imaging system. Therefore, the gel can be resolved further and scanned repeatedly (Figures 1C and 2). A near-infrared fluorescent imaging system primarily for Western blots was also tested to scan the gel, but only the advanced infrared imaging system was able to scan the gel with good resolution. The methodology described is specific to a particular infrared imaging software, although other software packages may be used.
- Clean the bed of the scanner with ddH2O and dry well before scanning. Wipe dry the glass plates of the gel and place the plates containing the gel on the scanner bed.
- Open the infrared imaging software and go to the 'Acquire' tab. When the thinner plate (1 mm) is placed on the scanner bed, use the settings of Channel: 700, Intensity: Auto, Resolution: 169 µm, Quality: medium, and Focus offset: 1.5 mm. When the thicker plate (3 mm) is placed on the scanner bed, use the settings of Channel: 700, Intensity: Auto, Resolution: 84 µm, Quality: medium, and Focus offset: 3.5 mm. Focus offset depends on the thickness of the glass plate.
- Select the area that the gel occupies on the scanner. Click 'Start' to begin the scan.
- Repeat electrophoresis and scan additionally as necessary.
Representative Results
Orange G loading dye (6x: 0.12 g Orange G in 100 mL 30% Glycerol) can be added to the binding reaction prior to loading to visualize the progress of the electrophoresis. Other loading dyes including bromophenol blue will be detected during scanning and therefore interfere with image analysis (Figure 1B).
In some instances of EMSAs, especially if nuclear extract preparation is used, poly deoxyinosinic-deoxycytidylic (dI-dC) is included in the binding reaction to decrease non-specific binding of proteins to DNA11. However, 50 µg/mL of dI-dC abolished the binding of purified 6xHis-SOX-2 with 5'Dye DNA probes (Figure 1B).
To show binding specificity of the infrared fluorescent dye-labeled DNA probes, we used WT or mutant cold probes as competitors (Figure 1C). In this case, the LIM-4(M)/SOX-2 cold probe mutated in the LIM-4 binding site is as efficient as WT LIM-4/SOX-2 cold probe in competing with infrared fluorescent dye-labeled LIM-4/SOX-2 probe for binding to 6xHis-SOX-2G73E (Figure 1C, lanes 3-6 vs. 7-10). However, the competition efficiency of the LIM-4/SOX-2(M) cold probe mutated in the SOX-2 binding site is much lower than WT LIM-4/SOX-2 cold probe for binding to 6xHis-SOX-2G73E (Figure 1C, Lanes 13-16 vs. 17-20).
The binding specificity of proteins can be determined by performing supershift assays of the protein-DNA interaction using antibodies specific for the proteins or tags, especially when nuclear extract preparation is used for the assay1. SOX-2 was tagged with 6xHis and FLAG epitopes. Addition of 1 µg anti-6xHis or 2.5 µg anti-FLAG M2 antibodies resulted in a supershifted SOX-2-DNA band (Figure 2).
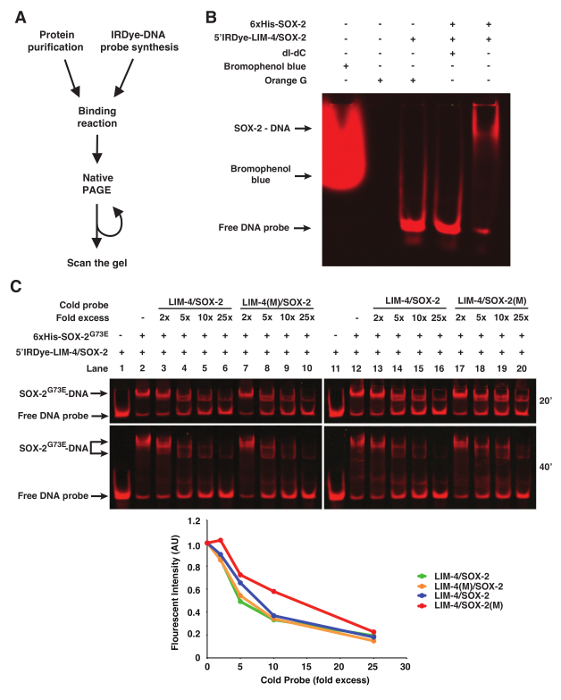
Figure 1. fEMSA using Infrared Fluorescent Dye-labeled Oligonucleotides. (A) Flow chart of fEMSA. (B) The effect of bromophenol blue dye, orange G, and dI-dC (1 µg) on fEMSA. 5 nM of 5'Dye-LIM-4/SOX-2 probe was used. (C) fEMSA showing binding specificity of 6xHis-SOX-2G73E to the SOX-2 target site of the LIM-4/SOX-2 element. 5 nM of 5'Dye-LIM-4/SOX-2 probe was used. LIM-4/SOX-2(M), mutant target sites. The gels were scanned at 20 and 40 min after electrophoresis. The scanned image obtained at 40 min after electrophoresis was used to quantify the fluorescent band intensity, which was normalized by the intensity of the lane without any cold probe. AU, arbitrary unit. Please click here to view a larger version of this figure.
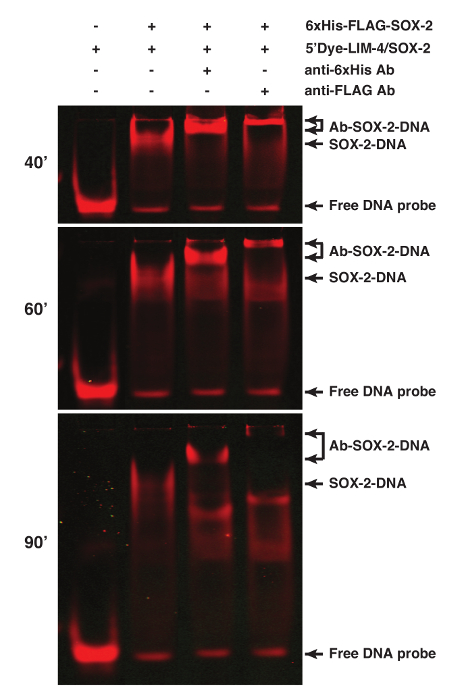
Figure 2. Supershift Analyses of the SOX-2-DNA Complex using fEMSA and Antibodies. SOX-2 was tagged with both 6xHis and FLAG epitopes. Addition of anti-6xHis or anti-FLAG antibodies caused a supershift of the SOX-2-DNA complex. 5'Dye-LIM-4/SOX-2 probe was used. Gel images were spliced between lanes 2 and 3 to exclude irrelevant lanes. The gel was scanned at 40, 60, and 90 min after electrophoresis. Please click here to view a larger version of this figure.
Discussion
fEMSAs are an efficient tool to investigate protein-DNA interactions, and are an alternative to radioactive labeling of DNA. Fluorescent dyes such as infrared dyes are commercially available, and provide a safer and more environmentally friendly method compared to radioactive DNA labeling. EMSAs using infrared fluorescent dye-labeled oligonucleotides do not require postrun gel processing steps, and therefore save time and cost compared to other DNA labeling techniques. The time to detect bands using radioactive EMSAs can range from 30 min to several days, depending on radioactivity and concentration of the DNA probes tested1. In contrast, scanning of fEMSA gels takes on average 25 min, thereby significantly reducing protocol time. fEMSAs also provide a significant advantage over radioactive labeling, as the gel does not have to be removed from the glass plates for analysis. This allows the gel to be rescanned if the initial run time is not sufficient to resolve protein-DNA complexes. The option to extend electrophoresis is not available using radioactive isotopes or biotin/streptavidin. Most importantly, handling radioactive material poses a safety risk to lab personnel, whereas fluorescent labeling of DNA probes is not hazardous, and can be disposed of easily. The use of streptavidin also requires a longer period until detection (approximately 3 h)12. The protocol and results presented here demonstrate that fEMSA is an effective and convenient tool for biomedical research. We have successfully used fEMSA to provide mechanistic insight into the effect of SOX-2G73E mutation on specific DNA binding activity that leads to olfactory neuron identity transformation5,10.
In the event that no or weak signal is detected when analyzing the gel, the concentration of DNA probe can be increased. The DNA probes used in our trials have been labeled with an infrared fluorescent dye on a single strand. While this method has worked successfully for probes we have tested, it has been reported that fluorescent labeling of a single strand has 30% intensity of probes that have been labeled on both strands. If intensity proves to be too low, it is recommended to label both strands of the DNA probe with an infrared fluorescent dye. If shifted bands are not observed, the protein-DNA complex may be unstable. Glycerol is crucial in this protocol to stabilize protein-DNA interactions. Glycerol is included in casted gels, the binding reaction, and the running buffer. Another point of consideration is choice of dye when tracking progression of samples through the gel during the run. Orange G is ideal for this purpose as other types of loading dyes such as bromophenol blue can interfere with scanning of the gel. Prerunning the gel is also important to ensure ammonium persulfate is removed from the gel prior to loading samples. In addition, use of poly dI-dC can interfere with binding of the protein of interest with the DNA probe in fEMSAs. Purity of the protein of interest and target DNA integrity are also important to their interaction. It is also important to ensure correct scanning settings when using the imaging software. The appropriate focus offset and intensity parameters are selected according to the thickness of the gel plates and intensity of DNA probes.
Previous protocols have been described to perform fluorescent EMSAs13,14. However, these protocols have required use of dI-dC, and/or pumps used to circulate water to maintain a particular temperature. The protocol described here provides a much more detailed method, and does not require use of any pumps.
Future applications for this protocol include monitoring competition assays in real time. Fluorescently labeled probes provide us with this additional insight, as gels can be scanned multiple times at different electrophoresis time points. In addition, DNA probes of interest and competitor probes can be labeled with different infrared fluorescent dyes, allowing for two-color imaging. As with all techniques, there are limitations to using fluorescent EMSAs. Higher concentrations of protein are needed as compared to traditional EMSAs due to relatively lower sensitivity. Another limitation is requirement of a specific imaging system with high resolution to detect the infrared fluorescent dye. Although the initial cost is high to purchase the scanner, long-term use of infrared fluorescent dye labeling is cost effective. Despite these limitations, fluorescent EMSAs provide an important method to address significant biological questions.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
This work was supported by an Alfred P. Sloan Research Fellowship (to C.-F.C.) and an NIH R01 grant (5R01GM098026-05 to C.-F.C.). We thank David Crowe for access to the advanced infrared imaging system.
Materials
| 30% Acrylamide/Bis Solution, 37.5:1 | Bio-Rad | 1610158 | Acrylamide is harmful and toxic. |
| 6x-His Epitope Tag Antibody (HIS.H8) | ThermoFisher | MA1-21315 | |
| Anti Flag M2 antibody | Sigma-Aldrich | F3165-.2MG | |
| Bovine Serum Albumin molecular-biology-grade | New England Biolabs | B9000S | |
| 5'IRDye700-labeled DNA Oligos | Integrated DNA Technologies | Custom DNA oligo | These are referred as 5'Dye-labeled or infrared fluorescent dye-labeled DNA oligos in the manuscript. The company will custom synthesize 5' IRDye labeled DNA oligonucleotides. Requires minimum 100μM scale synthesis and HPLC purification. |
| Klenow Fragment (3'–>5' exo-) | New England Biolabs | M0212S | |
| LightShif Poly (dI-dC) | ThermoFisher | 20148E | |
| Mini-PROTEAN Vertical Electrophoresis Cell | Bio-Rad | 1658000FC | This is referred as a mini protein gel system in the manuscript. Any electrophoretic system can be used as long as they are clear glass plates of less than 25cm x 25cm in size. |
| Odyssey CLx Infrared Imaging System | LI-COR Biotechnology | Odyssey CLx Infrared Imagng System | This is referred as an advanced infrared imaging system in the mansucript. |
| Odyssey Fc Imaging System | LI-COR Biotechnology | Odyssey Fc Dual-Mode Imaging System | This is referred as a near-infrared fluorescent imaging system primarily for Western blots in the mansucript. |
| Image Studio software (version 4.0) | LI-COR Biotechnology | Image Studio software | This is referred as a particular imaging software in the mansucript. |
| Orange G | Sigma-Aldrich | O3756-25G | |
| 6x Orange loading dye | 0.25% Orange G; 30% Glycerol | ||
| 0.5x TBE | 45 mM Tris-Borate; 1 mM EDTA | ||
| 1x TE | 10 mM Tris-HCl; 1 mM EDTA, pH8.0 | ||
| 1x STE | 100 mM NaCl; 10 mM Tris-HCl, pH8.0; 1 mM EDTA | ||
| 5x Binding Buffer | 50 mM Tris-HCl, pH7.5; 50 mM NaCl; 200 mM KCl; 5 mM MgCl2; 5 mM EDTA, pH8.0; 5 mM DTT; 250 ug/ml BSA |
Referencias
- Hellman, L. M., Fried, M. G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2, 1849-1861 (2007).
- Ludwig, L. B., Hughes, B. J., Schwartz, S. A. Biotinylated probes in the electrophoretic mobility shift assay to examine specific dsDNA, ssDNA or RNA-protein interactions. Nucleic Acids Res. 23, 3792-3793 (1995).
- Engler-Blum, G., Meier, M., Frank, J., Muller, G. A. Reduction of background problems in nonradioactive northern and Southern blot analyses enables higher sensitivity than 32P-based hybridizations. Anal Biochem. 210, 235-244 (1993).
- Jing, D., Agnew, J., Patton, W. F., Hendrickson, J., Beechem, J. M. A sensitive two-color electrophoretic mobility shift assay for detecting both nucleic acids and protein in gels. Proteomics. 3, 1172-1180 (2003).
- Alqadah, A., et al. Postmitotic diversification of olfactory neuron types is mediated by differential activities of the HMG-box transcription factor SOX-2. EMBO J. 34, 2574-2589 (2015).
- Jullien, N., Herman, J. P. LUEGO: a cost and time saving gel shift procedure. Biotechniques. 51, 267-269 (2011).
- Poulin-Laprade, D., Burrus, V. Electrophoretic Mobility Shift Assay Using Radiolabeled DNA Probes. Methods Mol Biol. 1334, 1-15 (2015).
- Ruscher, K., et al. A fluorescence based non-radioactive electrophoretic mobility shift assay. J Biotechnol. 78, 163-170 (2000).
- Pagano, J. M., Clingman, C. C., Ryder, S. P. Quantitative approaches to monitor protein-nucleic acid interactions using fluorescent probes. RNA. 17, 14-20 (2011).
- Alqadah, A., Hsieh, Y. W., Chuang, C. F. Sox2 goes beyond stem cell biology. Cell cycle. , (2016).
- Larouche, K., Bergeron, M. J., Leclerc, S., Guerin, S. L. Optimization of competitor poly(dI-dC).poly(dI-dC) levels is advised in DNA-protein interaction studies involving enriched nuclear proteins. Biotechniques. 20, 439-444 (1996).
- Sorenson, A. E., Schaeffer, P. M. In-gel detection of biotin-protein conjugates with a green fluorescent streptavidin probe. Anal. Methods. 7, 2087-2092 (2015).
- Onizuka, T., Endo, S., Hirano, M., Kanai, S., Akiyama, H. Design of a fluorescent electrophoretic mobility shift assay improved for the quantitative and multiple analysis of protein-DNA complexes. Biosci Biotechnol Biochem. 66, 2732-2734 (2002).
- Steiner, S., Pfannschmidt, T. Fluorescence-based electrophoretic mobility shift assay in the analysis of DNA-binding proteins. Methods Mol Biol. 479, 273-289 (2009).
