Design of the HTS assay
Two important factors were taken into consideration when designing the herein fluorescent assay. First, we needed to replicate a physiological condition in which T- or B cell activation would represent an ailment (e.g. graft-versus-host disease). Second, the assessment of cellular activation should be performed using a sensitive and quantitative method. Fluorescence is nowadays one of the primary choice for HTS read-outs as it corresponds to these needs.10,16 As a robust GFP read-out can be obtained following Nur77GFP-derived T or B cell stimulation both in vitro or in vivo,12 our assay exploited this strategy to differentiate between non-activated and activated cells as a working model for the identification of immunomodulatory compounds.
Originally, the design of our assay was based on GFP assessment by flow-cytometry using unfractionated stimulated splenocytes. In this case, both non-activated/activated T and B cells are stained with anti-CD3 or anti-CD19, respectively prior to GFP assessment. As shown in Figure 1A, T cells (CD3+ cells) represent approximately 30% of a given mouse spleen. At steady state, the GFP level was in the range of 10% (most likely due to post-thymic selection through the TCR during development or homeostatic stimulation in the periphery). Following TCR stimulation using the CD3/CD28 beads, a five to six-fold increase in the percentage of GFP expression (57% instead of 10%) was detected in CD3+ T cells (Figure 1A–B). Likewise, B cells (CD19+ cells) represent 50-55% of total splenocytes and their basal GFP expression is comparable to non-activated T cells (e.g. ~10%, Figure 1C). Interestingly, however, BCR stimulation using anti-mouse IgG/IgM and recombinant CD40L triggered a trivial increase in GFP expression (33% instead of 12%, Figure 1C–D). As GFP expression is a key element for assessing the pharmacological effect of screened small molecules on activated T or B cells, optimizing its intensity is central for the sensitivity and success of the screening.
Assessment of GFP expression using purified T or B cells
Although T and B cell stimulation can be achieved directly using unfractionated splenocytes, the GFP expression intensity was not sensitive enough for the screening especially if read-outs are to be performed by microscopy. In order to improve the cellular response, T cells were first purified by magnetic beads using a commercially available kit (Figure 2A). As previously shown using unfractionated splenocytes, 10% of isolated CD3+ T cells were GFP positive (basal expression). In this context, T cell stimulation using the CD3/CD28 beads significantly improved their GFP response (Figure 2A–2B, 86% as opposed to 57% when unfractionated splenocytes were used). Similar improvements were obtained with B cells (Figure 2C–2D, 80% instead of 33%). These results clearly demonstrate that the use of purified T and B cells maximizes GFP expression, which is suitable for the proposed phenotypic screening.
Schematic diagram of the designed HTS assay
Overall, the assay can be sub-divided into four major sections (Figure 3). In the case of T cells for example, a splenocytes cell suspension is prepared in order to purify T cells (steps 1-3) followed by a 12 h stimulation step in vitro using CD3/CD28 beads (steps 4-5). Activated T cells are then plated and cultured for 24 h with the small molecules of choice in 384-well plates (steps 6-7) before the assessment of GFP and Hoechst intensities using the automated HCS system. The same assay could be used for B cells with modifications performed at steps 4-5. Briefly, B cells can be stimulated with anti-mouse IgG/IgM and recombinant CD40L instead of beads. As these components are used in their soluble form (e.g. not linked to beads), B cells can be incubated for 12 h then simply washed prior to plating in 384-well plates for the screening process.
Efficient T-cell survival is primordial for the success of the HTS study. Accordingly, two limiting factors may impede the quality of the assay and should be taken into consideration. First, murine T cells are highly susceptible to apoptosis following in vitro stimulation.17 Second, all tested compounds are diluted in a DMSO solution, which might add a second stress factor. To minimize cell loss, recombinant IL-7 (2 ng/ml) was supplemented to T cells upon TCR stimulation at steps 4-5. This homeostatic cytokine supports proliferation and enhances T-cell survival through the expression of anti-apoptotic molecules such as Bcl-2, Bcl-xL and Mcl-1.18,19,20,21,22
Primary high throughput screening of 4,398 compounds reveals several activators and inhibitors of T cell activation
A high throughput screening was carried out by plating 75,000 of non-activated (negative control) or activated T cells (positive control) as displayed in Figure 4. To ensure high reproducibility, the assay was conducted several times with an obtained Z factor of 0.87 (Figure 5A). Using the HCS system, the observed GFP expression of stimulated T cells was 20 times higher than unstimulated T cells allowing the generation of a ''window of action'' zone where GFP inhibitory compounds could be easily detected (Figure 5A). This is shown in the figure in terms of the number of fluorescent cells divided by the total number of cells in the scanned fields. In the example shown in Figure 5A, the screening of 320 compounds unveiled three compounds (pointed-out by red arrows) capable of inhibiting GFP expression by 1.5-2.5 folds. In this example, the positive control value was 0.44 ±0.02 and the negative control was 0.02 ±0.00.
The screening of a total of 4398 compounds (Z factor of 0.85) was then conducted. The compounds were selected by medicinal chemists as seed/representative compounds based on their chemical structure representing an overall chemical library containing 136,000 entities (Figure 5B). At the end of the assay, analysis of GFP inhibition identified compounds capable of either inhibiting (positive deviators) or enhancing GFP expression (negative deviators).
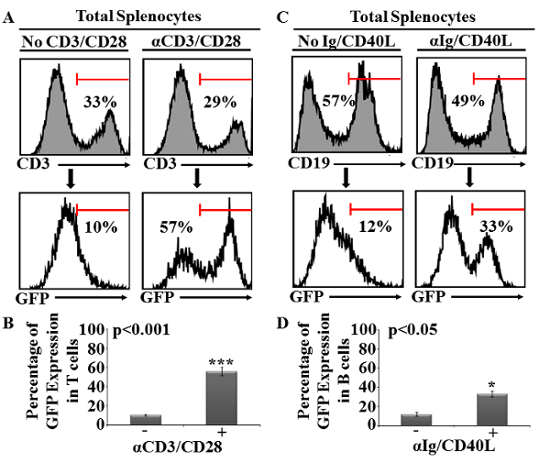
Figure 1: Analysis of T- and B-cell GFP expression following TCR or BCR activation conducted on unfractionated splenocytes. (A) Representative flow-cytometry analysis of T cells stained with an anti-CD3 antibody using splenocytes in the absence (top left panel) or presence of CD3/CD28 beads (top right panel). GFP expression for both T-cell groups are displayed at the bottom. (B) Percentage of GFP expression obtained by flow-cytometry analysis of T cells. Error bars represents mean ± SD. (C) Similar to (A) except that splenocytes were activated using anti-mouse IgG/IgM and recombinant CD40L then stained for CD19. (D) Percentage of GFP expression obtained by flow-cytometry analysis of B cells. Error bars represents mean ± SD. All shown experiments were repeated at least three times (n = 5/group; *P <0.05 and ***P <0.001). Please click here to view a larger version of this figure.
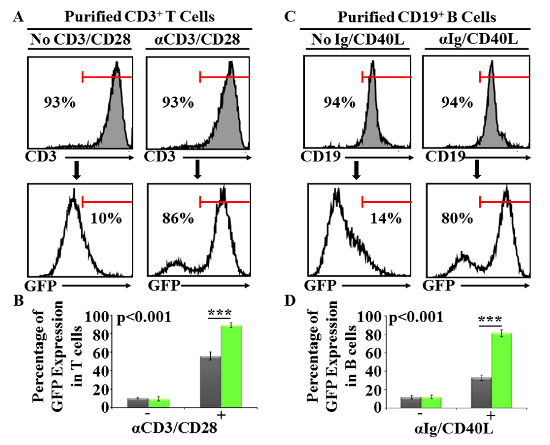
Figure 2: Analysis of GFP expression using purified T or B cells following TCR or BCR activation respectively. (A) Representative flow-cytometry analysis of purified T cells stained with an anti-CD3 antibody. For GFP induction, purified T cells were stimulated using CD3/CD28 beads (top right panel). Non-activated control T cells are shown at the top left panel. GFP expression of both non-activated and activated T cells is displayed at the bottom. (B) Percentage of GFP expression obtained by flow-cytometry analysis of T cells. Error bars represents mean ± SD. (C) Similar to (A) except that the purity of B cells was assessed by CD19 staining and activated using anti-mouse IgG/IgM and recombinant CD40L. (D) Percentage of GFP expression obtained by flow-cytometry analysis of B cells. Error bars represents mean ± SD. All shown experiments were repeated at least three times (n = 5/group and ***P <0.001). Please click here to view a larger version of this figure.
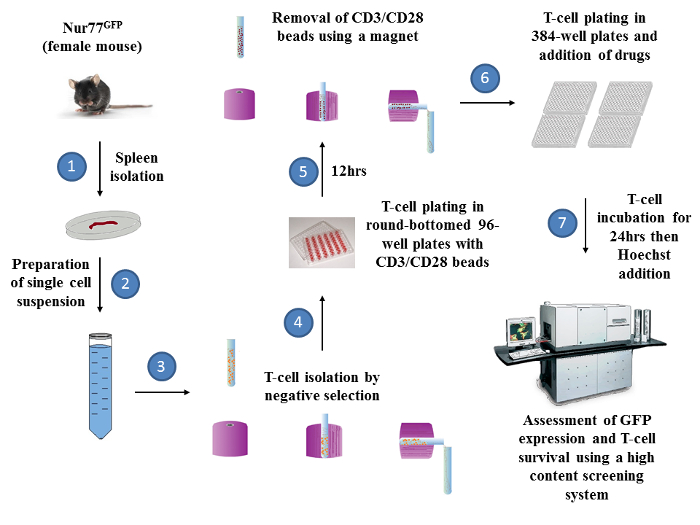
Figure 3: Overall schematic diagram representing the steps of the phenotypic screening. There are 7 major steps required to prepare the HTS assay. A female Nur77GFP mouse is sacrificed to isolate its spleen (Step 1). Following the generation of a splenocytes cell suspension (Step 2), T cells are negatively isolated (by depletion of unwanted cells) using a commercially available kit (step 3). Isolated T cells are then incubated with recombinant IL-7 in the absence or presence of CD3/CD28 beads in round-bottomed 96-well plate at 37 ºC (step 4). Twelve hours later (step 5), T cells are pipetted several times to dislodge any aggregates prior to beads removal using the same cell isolation magnet used originally for T-cell purification. Isolated T cells are then seeded in 384-well plates (75 000 cells/well, step 6) with the compounds for another 24 h (step 7). The following day, incubated cells are stained with Hoechst 30 min prior to analysis by the HCS system (step 8). The same set-up could be followed-up for the preparation of a B cell-based assay except that step 4 and 5 are to be replaced by a simple wash as the stimulators are added in soluble forms. Please click here to view a larger version of this figure.
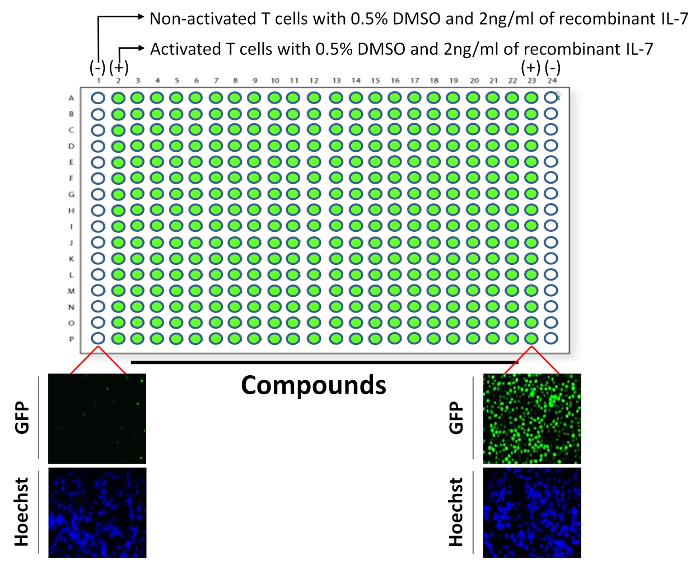
Figure 4: Layout for the plating of T cells in a 384-well plates. Each plate used in the assay contained three groups. In the first group, wells were loaded with non-activated T cells (negative control for GFP on each sides of the plate; wells A1-P1 and A24-P24), whereas the second group contained activated T cells without drugs (positive control for GFP; wells A2-P2 and A23-P23). All remaining wells containing activated T cells supplemented with the chemical entities used in the screening. All wells contained 0.5% DMSO. Examples for the GFP and Hoechst signals are shown for non-activated and activated T cells. Please click here to view a larger version of this figure.
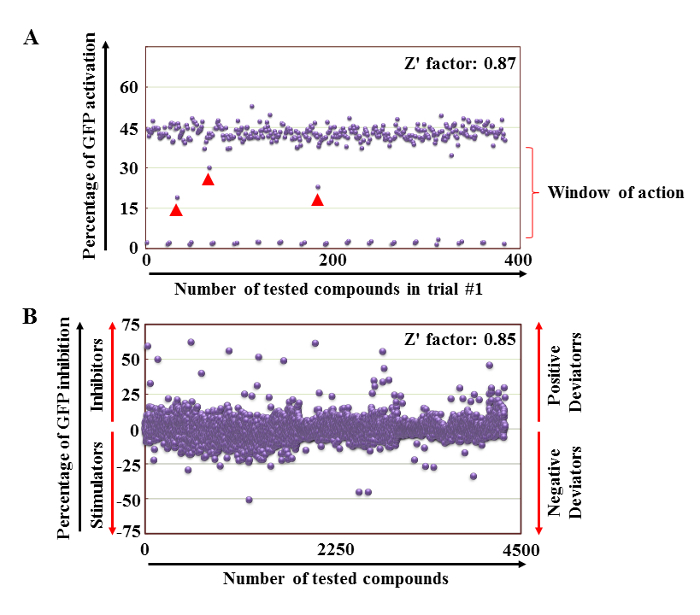
Figure 5: Primary phenotypic screening. (A) Representative example of a primary screening performed on 320 compounds. Non-activated T cells are almost GFP null in contrast to TCR-stimulated T cells, which display 20-fold higher GFP expression. (B) Cumulative data of all compounds screened using the assay. The GFP expression demonstrates compounds displaying inhibitory action whereas other compounds were behaving as T cell stimulators. Please click here to view a larger version of this figure.
