Highly Multiplexed, Super-resolution Imaging of T Cells Using madSTORM
Summary
We demonstrate a method to image multiple molecules within heterogeneous nano-structures at single molecule accuracy using sequential binding and elution of fluorescently labeled antibodies.
Abstract
Imaging heterogeneous cellular structures using single molecule localization microscopy has been hindered by inadequate localization precision and multiplexing ability. Using fluorescent nano-diamond fiducial markers, we describe the drift correction and alignment procedures required to obtain high precision in single molecule localization microscopy. In addition, a new multiplexing strategy, madSTORM, is described in which multiple molecules are targeted in the same cell using sequential binding and elution of fluorescent antibodies. madSTORM is demonstrated on an activated T cell to visualize the locations of different components within a membrane-bound, multi-protein structure called the T cell receptor microcluster. In addition, application of madSTORM as a general tool for visualization of multi-protein structures is discussed.
Introduction
A variety of super-resolution microscopy techniques have been developed to overcome the diffraction limit of light microscopy (~200 nm). Among these is a category of techniques called single molecule localization microscopy (SMLM) which includes photo-activation localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM). SMLM techniques share in the use of fluorophores that can be switched between on (fluorescent) and off (dark/photo-switched) states, allowing sequential localization of fluorescence from single molecules1,2,3.
Due to its compatibility with commercially available dyes and microscopes, direct STORM (dSTORM) has become a widely adopted SMLM technique4. dSTORM can routinely achieve ~10 nm localization precision, defined as the uncertainty in calculating the center of a diffraction-limited point spread function (PSF). However, despite the high precision estimated using localization algorithms5,6,7, accurate determination of the actual location of single molecules has been hampered by a number of issues. First, mechanical movement of the microscope stage during image acquisition adds significant uncertainty to localization precision. As SMLM images are obtained over thousands of time-lapse frames, nano-scale movements of the microscope stage can significantly compromise the precision of the final super-resolution image8. To compensate for stage movement during image acquisition, stage drift is commonly estimated from regression-based fitting of binned localizations from the image itself (cross correlation) or sequential localizations from fiducial markers (fiducial correction)1,9. However, these methods require optimization of multiple parameters for each image stack, and cannot account for stage movements at short time scales such as mechanical vibration. Gold nano-particles and multi-color fluorescent beads have been used as fiducial markers in SMLM, but they are not photo-stable, and result in significantly lower precision after drift correction than the nitrogen vacancy-center fluorescent nano-diamonds (FNDs) used in madSTORM10.
In addition to the diffraction limit, light microscopy is further restricted by spectral limits. Simultaneous visualization of multiple targets requires fluorescent probes with non-overlapping spectral profiles, generally restricting fluorescence-based light microscopy to 6 colors and SMLM to 2-3 colors4,11,12. Moreover, non-linear chromatic aberration causes misalignment of multicolor images, which require extensive alignment procedures using multi-colored fiducial markers8,13. To overcome these limits, previous studies have imaged multiple targets using repetitive photobleaching or chemical quenching of sequentially bound fluorophores14,15,16,17,18,19. While these methods can overcome the spectral limits of microscopy, fluorescence bleaching is known to be a toxic process20, and prolonged bleaching or quenching may cause unwanted side effects such as loss of crosslinking. Furthermore, the accumulation of fluorescent probes could lead to steric blocking of binding sites in the sample, preventing large-scale multiplexing and robust targeting of epitopes. To avoid such steric interference, a recent study achieved multiplexing using stochastic exchange of freely diffusing protein fragments21. Whereas this method allows dense labeling of cellular structures, it requires extensive biochemical preparation to isolate peptide fragments, cannot locate single molecule positions, and does not readily facilitate large-scale multiplexing using commercially available probes. We present a detailed video protocol describing the sequential binding and elution of fluorescently antibodies for multiplexed, antibody size-limited dSTORM (madSTORM) imaging, and use of fluorescent nano-diamonds to achieve precise drift correction and alignment.
Protocol
Caution: Please consult all relevant material safety data sheets (MSDS) before use. Several of the chemicals used in this protocol are toxic and carcinogenic. Please use all appropriate safety practices when performing the protocol including the use of engineering controls (fume hood, glovebox) and personal protective equipment (safety glasses, gloves, lab coat, full length pants, closed-toe shoes).
1. Multiplexed Imaging of Activated T Cells
- Directly conjugate antibodies with Alexa-647 (A647) dye using the Alexa 647 antibody labeling kit. Follow the labeling protocol provided by the manufacturer.
NOTE: Dialysis of antibody solution in 1x PBS is recommended before this step to increase antibody labeling efficiency. - Spin labeled antibodies for 5 min at 20,800 rcf and collect supernatant to remove aggregated antibodies.
- Preparation of fluorescent nano-diamond-coated coverslips
- Coat 8-well coverslip chambers (see reagent/material list) with 250 μL 0.01% Poly-L-Lysine (PLL) for 15 min, aspirate, and dry at 65 °C for 30 min. (No rinse before drying.)
- Prepare a dilution of 100 nm FNDs in 1x PBS. Test various dilutions to ensure enough FNDs are visible each field of view in step 1.2.7 (we are using a 1:200 dilution of FNDs provided by the manufacturer).
- Vortex the diluted FNDs for 1 min.
- Sonicate the FND supernatant for 30 s at a high-power setting.
- Incubate the sonicated FND supernatant in a PLL-coated 8-well coverslip chamber for 30 min at room temp.
- Wash 5x with 1x PBS and visualize the FND-coated chamber using 647 nm laser excitation on the TIRF microscope. Ideally, 4-10 individual FNDs should be visible in a field of view given the appropriate dilution of FNDs in step 1.2.2 (we are using a 100X objective with 256 x 256 camera pixel setting to yield 61 x 61 μm field of view) with at least one FND present in each quadrant of the imaging field. If the FNDs appear to be clustered (i.e. FNDs with significantly higher signal than surrounding FNDs and a point spread function larger than 200 nm), centrifuge FNDs at 6,800 rcf for 1 min, and repeat the coating procedure using the FND supernatant.
- Add 250 μl of anti-CD3 antibody (10 μg/mL) in each well and incubate for 1 hr at 37 °C, or overnight at 4 °C.
- Remove solution and add 1x PBS for 30 sec. Repeat this wash step 5 times (wash 5x).
- Activation of Jurkat T cells
- Spin 1 ml of Jurkat T cells at 800 rcf for 6 min and resuspend in 300 μl 1x HBS solution. Jurkat T cells should ideally be at a concentration of 0.5-1.0 x 106 cells/ml before spinning. 1x HBS solution consists of HEPES buffer saline with 1% Bovine serum albumin as described previously22.
- Add 150 μL of 1x HBS solution in each well and incubate at 37 °C for 15 min.
- Add 50 μL resuspended Jurkat T cells to each well and incubate for 3 min at 37 °C.
- Add 300 μL of 4% Paraformaldehyde to each well and incubate for 30 min at 37 °C.
- Wash 3x with 1x PBS.
- Permeabilize cells by adding 250 μL of 0.1% Triton-X solution for 5 min at room temp.
- Wash 3x with 1x PBS.
- Add 250 μL 1% fish gelatin solution in 1x PBS to each well for 30 min at room temp.
- Wash 3x with 1x PBS.
- Imaging activated Jurkat T cells using madSTORM
- Add 200 μL of labeled antibody at 0.1-0.5 μg/mL to fixed cells for 1 hr at room temp.
- Wash 5x with 1x PBS.
- Add 1 ml of STORM buffer and cover the chamber with a glass coverslip to limit exposure to air. The STORM buffer has been described previously10. We recommend making new STORM buffer for each round of imaging and spinning the STORM buffer at 20,800 rcf for 2 min to remove precipitates.
- Using low 647 nm laser power in TIRF mode, locate a stained cell with at least 3 FNDs in the field of view. To aid in locating FNDs, concomitant excitation with 568 nm laser can be used to increase the brightness of the FND signal.
NOTE: The performance of averaged fiducial correction described in section 2.1 improves with inclusion of more FNDs. - Increase 647 nm laser power and acquire images. We typically acquire 10,000 frames at 200 ms exposure, 125 mW 647 nm laser, 100X TIRF objective lens (1.49 NA), 100X EM gain (5 MHz at 16 bit, conversion gain 1). Details of our imaging setup have been described previously10.
- Wash 5x with 1x TBS.
- Add 1 ml of elution buffer (3.5 M MgCl2, 20 mM PIPES, 0.1% Tween-20, pH 6.5) and incubate at room temp for 1 min.
- Repeat elution 3 times. (exact elution conditions and the number of elution rinses needed to remove the signal must be checked for each antibody used).
- Wash 3x with 1x TBS and add 1 ml of 1x PBS.
- Photobleach using 647 nm laser at high power (125 mW) with 405 nm laser at 2-5 mW to photo-activate A647 dyes in dark state. Wait until all remaining signal from non-eluted antibody is photobleached. Typically this takes 2-5 s of laser exposure. Acquiring sample images (~100 frames) using the STORM settings in 1.3.5 after elution and photobleaching is recommended to confirm removal of signal.
We typically remove 99.8% of signal using this method. We recommend testing the elution efficiency of each antibody before use in madSTORM as shown previously10. - Add 250 μL 4% Paraformaldehyde for 15 min. This prevents reverse crosslinking of fixed molecules in the cell.
- Wash 3x with 1x PBS.
- Repeat steps 1.3.1-1.3.12 for sequential labeling of multiple targets. Figure 1 shows a composite image of multiple molecules in an activated Jurkat T cell acquired using madSTORM.
2. Drift Correction and Alignment of Multiplexed Image Stacks
- Drift correction of madSTORM image stacks
- Open images using ImageJ. We recommend converting the image files to a TIFF format before opening in ImageJ. Use rectangle ROI to select a single FND. Play the time lapse to make sure that the FND is visible in every frame of the image stack. Use Run Analysis in the ThunderSTORM plugin to localize the FND. For the averaged fiducial correction method described in 2.1.5, it is important that a single FND is localized in each image frame (i.e. number of identified peaks should be equal to the number of frames).
- If the FND is not identified in every frame, select another FND. If multiple peaks are located in a single frame, remove the peak that deviates the most from previous peaks.
- Repeat 2.1.2 for each FND in the image stack. Keep in mind that some FNDs should not be included in the drift correction process so they can serve as an independent gauge for drift correction precision as described in step 2.1.7.
- For faster, less precise drift correction, use either the cross correlation or fiducial correction algorithms included in ThunderSTORM to correct the FND images. Typically, we use 100 bins, 5 magnification, and 0.1 trajectory smoothing factor for cross correlation and 10 max distance, 0.1 min marker visibility ratio, and 0.01 trajectory smoothing factor for fiducial correction. Cross correlation and fiducial correction will yield a correction file which can be applied to other FNDs to test the drift correction precision.
- For more stringent, more precise drift correction, use the averaged fiducial correction (AFC) algorithm as described previously10. This process does not require optimization of parameters and yields higher localization precision than predicted by SMLM localization algorithms. However, averaged drift correction requires at least 4 FNDs and for the FNDs to be localized in every frame of the image stack. AFC drift correction performs better with inclusion of more FNDs because a large number of localized FNDs will lessen the distorting effect of an outlier peak in a given frame.
- Localize all point spread functions within the image stack using ThunderSTORM as described previously10. We typically use pixel size of 100 nm, photoelectrons per A/D count of 4.28, base level A/D count of 100, EM gain of 100, PSF: Integrated Gaussian localization, 5 pixel fitting radius, maximum likelihood fitting method, and 1.3 pixel initial sigma.
- Apply the drift correction factor acquired from FNDs to localizations from the entire image stack. Visually inspect the final image to ensure proper drift correction (Figure 2). The resulting drift correction factor should be independently tested on FNDs not included in the AFC procedure to measure the level of drift correction precision. The precision of drift-corrected, independent FNDs should be close to the average precision/uncertainty value calculated from the localization algorithm utilized in step 2.1.6.
- Repeat steps 2.1.1-2.1.7 for the madSTORM image stacks of sequentially labeled antibodies. Keep in mind, that localizing the same set of FNDs in each madSTORM image stack is critical for the following alignment procedure.
- Alignment of madSTORM images
- Calculate the centroid position of the drift corrected FNDs in each madSTORM image. To do this, find the mean x and y axis positions for each localized FND, and average the mean x and y positions of all FNDs in each madSTORM image. The detailed algorithm for this step has been described previously10. Align sequential madSTORM images using the centroid positions of FND fiducial markers. To perform the alignment, calculate the offset between two localized madSTORM images by subtracting the centroid position of FNDs in the second madSTORM image from the first. Then subtract the offset amount from all localizations in the second madSTORM image.
- We recommend using the first madSTORM image as a reference image and to repeat this alignment step between the first and third, first and fourth, etc. Test the alignment precision by measuring the offset between FNDs not included in the alignment process. Large offsets may indicate that different sets of FNDs were used when calculating the centroid positions of sequential madSTORM images.
- If so, steps 2.2.1-2.2.2 should be repeated using the same set of FNDs to perform alignment.
NOTE: Custom codes for both the AFC drift correction and alignment procedures are provided.
Representative Results
The sequential elution and staining method was used to produce the multiplexed madSTORM image of microclusters and other structures in an activated Jurkat T cell (Figure 1, check figure alignments). Each pseudo-colored image represents one round of madSTORM imaging acquired in steps 1.1.1 to 1.3.13. As described previously10, the field of view should be monitored for residual signal from previous, non-eluted antibody to avoid crosstalk between multiplexed targets. Moreover, as the elution efficiency can vary among antibodies, the order of sequential multiplexing should be arranged from the best to worst eluting antibody to minimize steric blocking of epitopes. The final madSTORM image in Figure 1 has been corrected for drift and alignment using AFC and FND fiducial markers as described in section 2.
FNDs were used to perform AFC on localizations from a single, independent FND (Figure 2A, 2B) and an activated Jurkat T cell (Figure 2C, 2D). The AFC drift correction was performed as described in steps 2.1.1-2.1.8 using the algorithms provided. First, visual inspection of the image after drift correction (Fig. 2C) is recommended to ensure proper application of the drift correction algorithm. Second, measuring the standard deviation of localizations in both the X and Y axis is recommended to confirm the precision achieved by AFC. As reported previously10, AFC typically yields better precision than expected by various localization algorithms.
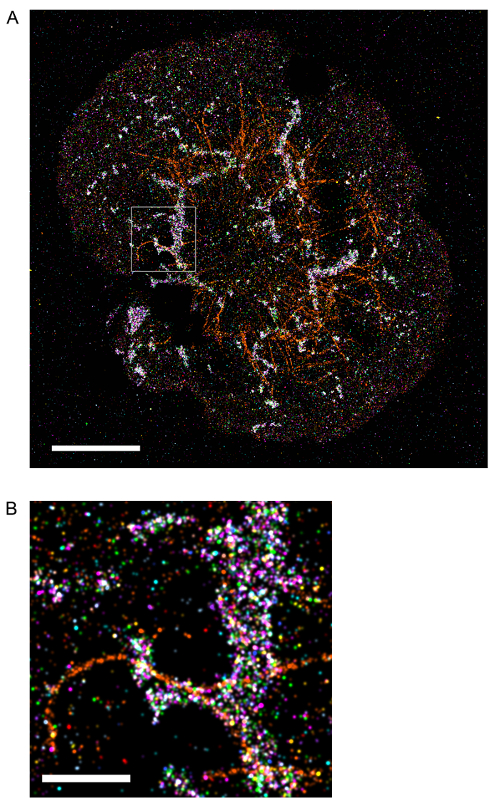
Figure 1: (A) An activated Jurkat T cell, sequentially imaged using multiple rounds of madSTORM imaging. LAT (red), pLAT-Y171 (magenta), pLAT-Y191 (pink), pLAT-Y226(purple), PLCγ1 (light blue), pSLP76-Y128 (green), pSRC-Y416 (aqua), pTCRζ-Y142 (blue), TOM20 (yellow), αTubulin (orange), ZAP70 (brown). Scale bar = 2.5 μm. (B) Expanded image of boxed region in A. Scale bar = 500 nm. Please click here to view a larger version of this figure.
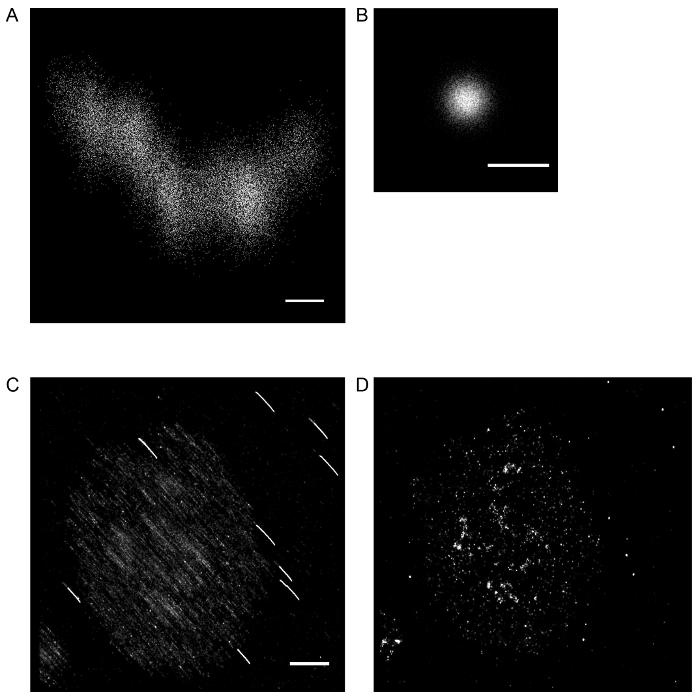
Figure 2: SMLM image of a single FND localized in 30,000 image frames (A) before and (B) after drift correction with averaged fiducial correction. Scale bars = 20 nm (A and B). SMLM image of an activated Jurkat T cell stained with anti-phosphorylated SLP76 (Y128) antibody and FND fiducial markers (C) before and (D) after drift correction with averaged fiducial correction. Scale bar = 2 μm. Please click here to view a larger version of this figure.
Discussion
The sequential multiplexing, drift correction, and alignment procedures in madSTORM allow precise, highly multiplexed visualization of heterogenous structures in cells.10 In addition, madSTORM avoids the limitations of multi-color STORM such as chromatic aberration and sub-optimal photoswitching/emission properties of non-far red dyes9,12. As the elution step significantly reduces steric interference from sequentially bound antibodies, madSTORM can be used to perform repeated imaging of cell samples beyond the 6-10 rounds of multiplexed imaging achieved by previous techniques9,15,21. Moreover, the sequential elution and staining steps are not restricted to SMLM and can be used for other imaging techniques such as confocal microscopy, SIM and two-photon microscopy. However, given that madSTORM is a sequential immunostaining technique, this technique is limited by the availability of antibodies for each targeted molecule. Each antibody should be carefully tested to ensure specificity, labeling efficiency, and elution ability.
The acquisition settings for madSTORM were chosen to maximize detection of individual photoswitching events, allowing us to achieve 2.5 nm average localization precision. However, the high precision comes at the expense of time, requiring ~3 h for each round of madSTORM imaging as discussed previously10. For faster image acquisition at lower localization precision (5-10 nm), a shorter exposure time and higher EM gain are recommended (e.g. 20 ms exposure, 300X EM gain, 17 MHz at 16 bit, conversion gain 3). Furthermore, multi-color imaging (e.g. Atto-488, Alexa-568, and Alexa-647 nm) can be performed for each round of madSTORM imaging to increase the scale of multiplexing, albeit with a decrease in alignment precision due to chromatic aberration.
We have successfully performed up to 25 rounds of madSTORM imaging using this protocol. As the entire procedure is performed with the coverslip chamber mounted on the microscope stage, it is important to secure the coverslip chamber in the same position. To do this utilize auto focus mode and mount the chamber on the microscope stage using metal clamps. If significant stage drift has occurred, use known positions of FND fiducial markers to re-center the cell sample.
Simultaneous visualization of aligned madSTORM images can be challenging. To simultaneously view 7 or fewer madSTORM images, use Color/merge channels in ImageJ to create a pseudo-colored composite image. For 8 or more images, it is advisable to catagorize the madSTORM images into different groups, merge each group into a composite image, and apply a single LUT color to each composite image.
We used 100 nm nitrogen vacancy-center nano-diamonds due to their brightness and adherence to PLL-coated coverslips. FNDs smaller than 80 nm did not adhere well to PLL-coated coverslips, and FNDs larger than 100 nm were significantly brighter than Alexa-647 dye when excited using 647 nm laser, limiting optimization of camera acquisition settings for Alexa-647 emission. We recommend experimenting with different FND sizes or colors if the 100 nm nitrogen vacancy-center FNDs are not optimal for your imaging setup. Alternatively, nano-gold particles or fluorescent beads can be used as fiducial markers, but are not optimal for the averaged point correction procedure used below in section 2.110. Finally, for NV– vacancy FNDs, the emission intensity can be decreased by up to ~50% in situ by the application of a static magnetic field23. The decrease in intensity is roughly linear up to ~500 Gauss at which point the effect saturates. In practice, the intensity of diamonds in the field of view can be decreased to the desired degree by bringing a permanent magnet closer to the microscope slide from the top.
Lastly, we optimized the elution buffer for removal of antibodies targeting the molecular components of the T cell microcluster. madSTORM is not limited to TCR microclusters, however, as microtubules, mitochondria, F-actin, Vimentin and other molecular structures have been imaged using this technique10. For antibodies targeting other cellular compartments, additional steps may be required to disrupt epitope binding such as higher temperature, lower pH and longer incubation of elution buffer. Conversely, the elution buffer can be used without Tween-20 to target sensitive membrane structures, preventing disruption of non-ionic interactions. We foresee in future experiments that the elution buffer composition will be tailored for individual antibodies to optimize elution efficiency and sample preservation. Furthermore, beyond the application in activated T cells, madSTORM may be feasible with other systems including in vitro molecular complexes, different cell types, and tissue sections. Various parameters of the madSTORM protocol, such as fixation, permeabilization, antibody staining, and elution buffer composition will need to be adjusted for each targeted system.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
We thank Xufeng Wu for access to the STORM microscope. This research was supported by the Intramural Research Program of the National Cancer Institute (NCI) Center for Cancer Research and the National Heart Lung and Blood Institute (NHLBI).
Materials
| 8 well coverslip chamber | Lab-tek | 155409 | |
| 0.1% Poly-L-Lysine solution | Sigma-Aldrich | P8920 | |
| NV-100nm Fluorescent Nano-diamond | Adamas | Red FND | |
| anti-CD3ε antibody | BD Biosciences | 555329 | |
| Bovine serum albumin, Fraction V | KSE Scientific | 98-100P | |
| Triton-X | Sigma-Aldrich | T9284 | |
| 10% Paraformaldehyde | EMS | 15712 | Carcinogen |
| Gelatin from fresh water fish skin | Sigma-Aldrich | G7041 | |
| Alexa 647 antibody labeling kit | Thermo Fisher | A20186 | |
| Magnesium Chloride hexahydrate | Sigma-Aldrich | 138924 | |
| PIPES | Sigma-Aldrich | P6757 | |
| Tween-20 | Fisher Scientific | BP337-500 | |
| 2-Mercaptoethanol | Sigma-Aldrich | M7154 | Highly toxic, air sensitive |
| Cysteamine | Sigma-Aldrich | 30070 | Highly toxic |
| Cyclooctatetraene 98% | Sigma-Aldrich | 138924 | Highly toxic, air sensitive |
| 10x PBS | KD Medical | RGF-3210 | |
| 10x TBS | KD Medical | RGF-3385 |
Referencias
- Betzig, E., et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642-1645 (2006).
- Hess, S. T., Girirajan, T. P., Mason, M. D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 91 (11), 4258-4272 (2006).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 3 (10), 793-795 (2006).
- van de Linde, S., et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc. 6 (7), 991-1009 (2011).
- Mortensen, K. I., Churchman, L. S., Spudich, J. A., Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 7 (5), 377-381 (2010).
- Thompson, R. E., Larson, D. R., Webb, W. W. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 82 (5), 2775-2783 (2002).
- Rieger, B., Stallinga, S. The lateral and axial localization uncertainty in super-resolution light microscopy. Chemphyschem. 15 (4), 664-670 (2014).
- Pertsinidis, A., Zhang, Y., Chu, S. Subnanometre single-molecule localization, registration and distance measurements. Nature. 466 (7306), 647-651 (2010).
- Bates, M., Jones, S. A., Zhuang, X. Stochastic optical reconstruction microscopy (STORM): a method for superresolution fluorescence imaging. Cold Spring Harb Protoc. (6), 498-520 (2013).
- Yi, J., et al. madSTORM: a superresolution technique for large-scale multiplexing at single-molecule accuracy. Mol Biol Cell. 27 (22), 3591-3600 (2016).
- Bates, M., Huang, B., Dempsey, G. T., Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 317 (5845), 1749-1753 (2007).
- Dempsey, G. T., Vaughan, J. C., Chen, K. H., Bates, M., Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 8 (12), 1027-1036 (2011).
- Erdelyi, M., et al. Correcting chromatic offset in multicolor super-resolution localization microscopy. Opt Express. 21 (9), 10978-10988 (2013).
- Gerdes, M. J., et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A. 110 (29), 11982-11987 (2013).
- Jungmann, R., et al. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods. 11 (3), 313-318 (2014).
- Nanguneri, S., Flottmann, B., Horstmann, H., Heilemann, M., Kuner, T. Three-dimensional tomographic super-resolution fluorescence imaging of serially sectioned thick samples. PLoS One. 7 (5), e38098 (2012).
- Schubert, W., et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 24 (10), 1270-1278 (2006).
- Tam, J., Cordier, G. A., Borbely, J. S., Sandoval Alvarez, A., Lakadamyali, M. Cross-talk-free multi-color STORM imaging using a single fluorophore. PLoS One. 9 (7), e101772 (2014).
- Valley, C. C., Liu, S., Lidke, D. S., Lidke, K. A. Sequential superresolution imaging of multiple targets using a single fluorophore. PLoS One. 10 (4), e0123941 (2015).
- Hoebe, R. A., et al. Controlled light-exposure microscopy reduces photobleaching and phototoxicity in fluorescence live-cell imaging. Nat Biotechnol. 25 (2), 249-253 (2007).
- Kiuchi, T., Higuchi, M., Takamura, A., Maruoka, M., Watanabe, N. Multitarget super-resolution microscopy with high-density labeling by exchangeable probes. Nat Methods. 12 (8), 743-746 (2015).
- Campi, G., Varma, R., Dustin, M. L. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 202 (8), 1031-1036 (2005).
- Sarkar, S. K., et al. Wide-field in vivo background free imaging by selective magnetic modulation of nanodiamond fluorescence. Biomed Opt Express. 5 (4), 1190-1202 (2014).
