EVs were isolated from whole blood and characterized by nanoparticle tracking analysis with fluorescing reagents. The optimal sensitivity for measurement of unstained particles was identified to range at 70% during our experiments. The fluorescent beads used for adjustment and calibration of the measurement showed an optimum setting at a sensitivity of 85% (Figure 2A). Between a sensitivity of 70% and 90%, the number of detected particles increased rapidly, while further increasing the sensitivity can lead to a deterioration of the particle size distribution where the number of particles is re-dropping. The settings of the camera displayed a sharp picture (Figure 2B) and repeated measurements showed low standard deviation (Figure 2C). Insofar, the protocol for processing the samples of EVs was adjusted so that all measurements could be conducted with the same settings (Table 2). The distribution width is defined by three values on the x-axis, the x10, x50, and x90. The x50, or median particle size is the diameter at which half of the population lies below this value. Similarly, the x10 and x90 indicate the diameter at which 10% and 90% of the detected particles are under the reported size. Staining with PKH67 cell linker kit, including a fluorescent cell linker that incorporates a green fluorescent dye with long aliphatic tails into lipid regions of the cell membrane, demonstrated a strong correlation between the sensitivity and the number of particles measured (Figure 3A). PKH67 is often used for proliferation monitoring but has also proven useful for monitoring exosome or liposome uptake as well as for in vivo cell trafficking. Due to the non-specific labeling of PKH67, a wide variety of EVs can be labeled and detected. The distribution of the particles was in a range between 266 nm (x10) and 1946 nm (x90) with a peak maximum at 857 nm (x50) and with a low standard deviation between the measurements (28.1 nm). LAMP-1, also known as lysosome-associated membrane glycoprotein 1, and CD107a reside primarily across lysosomal membranes. After staining with an Alexa Fluor 488 labeled specific antibody against LAMP-1, the distribution of the particles ranges from 220 nm (x10) to 1145 nm (x90) with a peak maximum at 541 nm (x50) and a standard deviation of 11.7 nm (Figure 3B). For characterization of EVs, we used Alexa Fluor 488 labeled antibodies against common exosomal markers and confirmed our findings by Western blotting. After staining with Alexa Fluor 488-labeled CD9-antibody, the distribution of the particles ranges from 251 nm (x10) to 1139 nm (x90) with a peak maximum at 548 nm and a second minor peak at approximately 25 nm (Figure 4A). Staining with Alexa Fluor 488 labeled CD63 (Figure 4B) and vimentin (Figure 4C) yielded similar results. Western blotting analysis substantiated our positive result for antibodies used here. Repeated measurements showed reproducible results for all antibodies used in this report. As controls, we stained vesicle-free water with respective antibodies (Figure 5A), where PKH67 and LAMP1 antibodies virtually detected no EV up to a sensitivity close to 100%. Using the example of vimentin, high sensitivity increased the number of emerging artefacts, even when the sample is essentially free of particles. If measurement is started when the drift is yet too high (> 5 µm/s), the individual repetitions distinctly deviate among themselves (Figure 5B). As represented with three different antibodies, it is crucial that the drift is as minimal as possible before starting the measurement. According to our experience, using fluorescein isothiocyanate (FITC) as fluorochrome results in measurements that are not accurate and reproducible because FITC is prone to rapid photo bleaching (Figure 5C). Therefore, we recommend using exclusively Alexa Fluor 488 labeled antibodies for EV characterization. In this protocol, EVs were isolated by a polymer-based exosome precipitation solution containing polyethylene glycol. To ensure that our results are not falsified by the applied isolation method, we characterized EVs after isolation with ultracentrifugation. As represented with PKH67 and two different antibodies (CD63 and LAMP-1), the results of our applied isolation with exosome precipitation solution (Figure 6A) are comparable with EVs isolated via ultracentrifugation (Figure 6B). Unfortunately, because of the poor yield of EVs after ultracentrifugation, the initial set of serum for isolation must be distinctly higher compared to isolation with exosome precipitation solution.
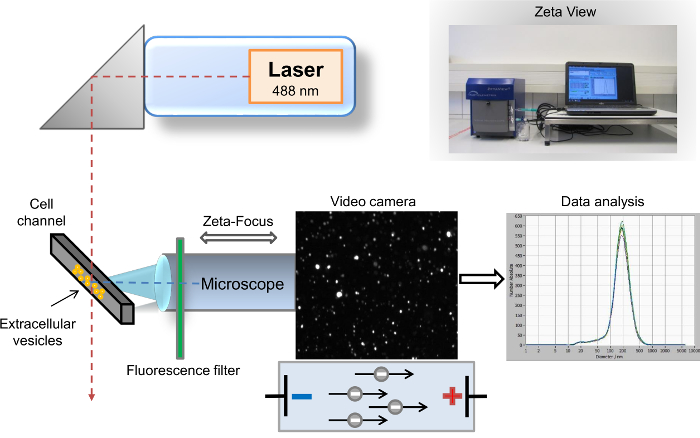
Figure 1: Schematic setup of the nanoparticle tracking analysis. The microscope/video axis and laser beam are orientated orthogonally to each other, crossing at the cell channel cross section. A fluorescence filter is placed between the cell channel and the microscope. Light scattered by the particles is displayed in the "live-view" window of the software. After acquisition, the results are displayed as a size distribution curve coordinate system. Please click here to view a larger version of this figure.
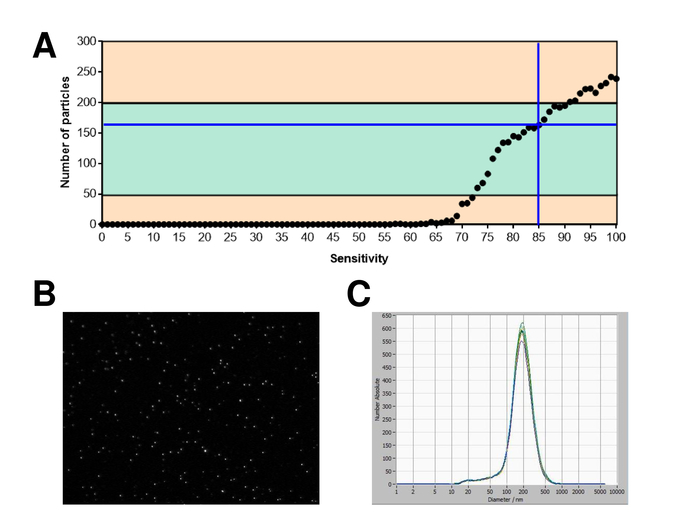
Figure 2: Calibration with fluorescence labeled beads. (A) The number of particles vs. sensitivity curve displays the particles in one position at one point in time during an automatic sensitivity scan. (B) Visualization of particles on the live view screen. (C) Particle size distribution after repeated measurements. Please click here to view a larger version of this figure.
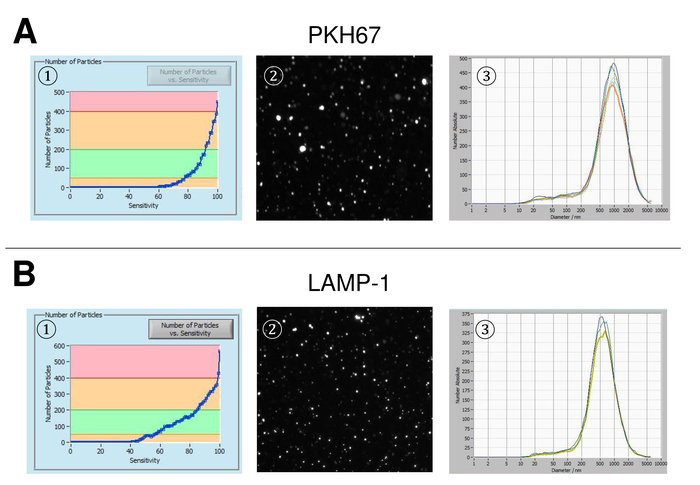
Figure 3: Representative results after staining with PKH67 and LAMP-1. Number of particles vs. sensitivity curve (1), visualization of particles on the live view screen (2), and particle size distribution (3) after staining with PKH67 (A) and LAMP-1 (B). A similar size distribution of particles is observed, while changes in the sensitivity setting lead to a similar but yet not entirely identical change of detected particles. Please click here to view a larger version of this figure.
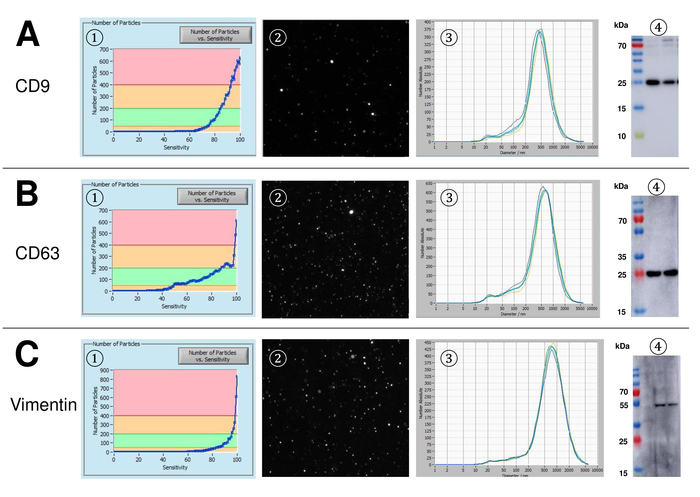
Figure 4: Representative results after staining with Alexa Fluor 488 labeled CD9, CD63, and vimentin. Number of particles vs. sensitivity curve (1), visualization of particles on the live view screen (2), particle size distribution (3), and representative Western blots of two different EV suspensions (4) for CD9 (24-27 kDa, A), CD63 (26 kDa, B), and vimentin (54 kDa, C). Late occurrence of particle signals along the increasing sensitivity (x-axis) correlates with lower signal intensity in the Western blot analysis, confirming the lower amount of the respective particle surface marker (e.g., vimentin vs. CD63). Note the almost identical curves of representative measurements for all analyzed markers indicating high reproducibility (3). Please click here to view a larger version of this figure.
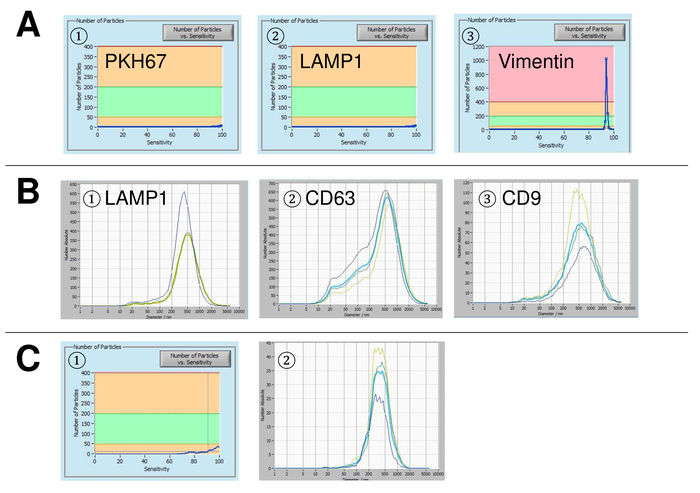
Figure 5: Representative results for used controls and possible error sources. (A) Vesicle-free water stained with PKH67 (1), LAMP-1 (2), and vimentin (3) as controls. (B) Representative particle size distributions after staining with LAMP-1 (1), CD63 (2), and CD9 (3), and measurements performed when suspension drift is yet too high. (C) Number of particles vs. sensitivity curve (1) and particle size distribution (2) after staining with FITC-labelled CD63 antibody. A clear bleaching effect is observed after each measurement, resulting in progressively lower particle numbers. Please click here to view a larger version of this figure.
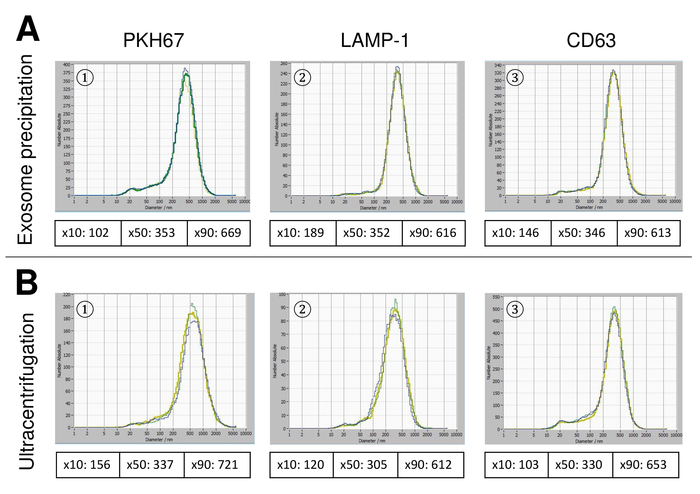
Figure 6: Comparison of different isolation methods for EVs. Particle size distribution of EVs isolated with exosome precipitation solution (A) and ultracentrifugation (B) after staining with PKH67 (1), LAMP-1 (2), and CD63 (3). Please click here to view a larger version of this figure.
| LAMP-1 | CD9 | CD63 | Vimentin | |
| Antibody (µL) | 5 | 2.5-5 | 2.5-5 | 5 |
| EV suspension (µL) | 10 | 20 | 10 | 10 |
| H2O (µL) for staining | 50 | 50 | 50 | 50 |
| Final volume (mL) | 5-10 | 2.5-5 | 5 | 10 |
Table 1: List of used antibodies and dilution range of samples.
| Acquisition parameters | |
| Sensitivity (%) | 85 |
| Shutter | 70 |
| Min. Brightness | 20 |
| Max. Size (nm) | 500 |
| Min. Size (nm) | 20 |
| Polarity | Negative |
| Voltage | Off |
| Particle drift at 0 V (µm/s) | < 5 |
| Positions | 11 |
| Cycles | 10 |
| Multiple acquisitions | 3-5 |
| Time delay (min) | 0 |
Table 2: Acquisition parameters for nanoparticle tracking analysis.
