Assessing the role of pathogen and host-encoded factors impacting cellular infection
Using the infection conditions described above, 0.15% of the input wild-type Lm is recovered after 1.5 h of co-incubation with cultured macrophages (Figure 1A). In the subsequent 1.5 h of co-incubation (3 h post infection, hpi), there was a 4-fold increase in recovery of viable Lm and from 3 to 6 hpi there was an additional 7.5-fold increase in the recovery of viable Lm. Since the cell-impermeable antibiotic gentamicin is added to the co-cultures at 1 hpi to eliminate extracellular Lm, the 30-fold increase in viable Lm levels between 1.5 and 6 hpi exclusively represents intracellular proliferation.
Lm rapidly gains access to the cytosol following its internalization within eukaryotic cells (see Introduction). Cytosolic Lm associates with the actin polymerization machinery by which means it propels itself capable of penetrating the plasma membrane. In the in vitro infection assay described above, if gentamicin is removed from the wells at 6 hpi, extracellular Lm can be readily detected 1 h later (7 hpi) in the overlying media (Figure 1B). No CFUs were recovered from control wells that were not seeded with macrophages but that were otherwise treated identically as seeded wells thereby showing that the appearance of extracellular Lm was entirely dependent on the presence of macrophages and not a result of incomplete antibiotic killing (not shown).
The capability of Lm to survive and proliferate intracellularly is largely dependent on a suite of genes whose expression are controlled by the transcription factor Positive Regulatory Factor A (PrfA)1. Initially, there is a comparable recovery of the wild-type Lm strain and a Lm mutant strain deleted for this factor (ΔprfA) (Figure 1A). However, there is a subsequent reduction in the recovery of the ΔprfA strain after prolonged co-incubation such that the recovery at 6 hpi is 3-fold less than the recovery at 1.5 hpi. Relatedly and expectedly, ΔprfA bacteria were not detected in the overlying media at 7 hpi (Figure 1B).
The activity of PrfA is regulated at multiple levels. A number of constitutively active PrfA variants, encoded by hyper-virulent prfA* alleles, have been isolated3. A Lm strain possessing a prfA* has an initial infection profile that is comparable to the isogenic wild-type strain at 1.5 and 3 hpi (Figure 1A). However, between 3 and 6 hpi, the fold-increase of the prfA* strain significantly differed from that of the wild-type strain. Again, relatedly and expectedly, the prfA* strain was readily detected in the overlying media at 7 hpi at levels that significantly exceeded that observed for the wild-type Lm strain (Figure 1B).
Following its release into the cytosol, Lm associates with the cellular actin polymerization machinery that results in the bacterium being enveloped by polymerized actin. To confirm that extracellular Lm is derived from the actin-rich cytosol of the infected host cell, macrophages were either left uninfected or infected with a GFP-expressing prfA* strain and the overlying media was collected at 1.5, 4, and 6 hpi, stained with phalloidin that binds polymerized actin, and analyzed by flow cytometry. Media overlaying both uninfected and infected macrophages contained light-scattering bacterium-sized particulate matter (Figure 2A), however, only media from infected macrophages possessed GFP-positive signals within this size gating (Figure 2B; left panel). There was a time-dependent increase in the appearance of phalloidin-positive Lm in the media overlaying infected macrophages (Figure 2B; right three panels). Phalloidin-positive GFP-expressing Lm were also detected in lysates prepared from infected macrophages (Figure 2C; left panel). However, no phalloidin-positive Lm was detected in lysates prepared from macrophages infected with a GFP-expressing Lm mutant strain that lacked the virulence factor ActA that is required to recruit the actin-polymerization machinery to the outer surface of Lm following its release into the cytosol (Figure 2C; right panel). These data support a scenario that in an in vitro infection model 'emergent' extracellular Lm are derived from the infected cell cytosol.
In addition to Lm-encoding factors, a number of host factors have been identified that impact Lm cellular infection (see Discussion). Recently, the host factor Perforin-2 (P2) has been shown to be a critical component of cell-level defenses against a number of bacterial pathogens. Peritoneal exudate macrophages (PEMs) isolated from wild-type (P2+/+) and P2-/- mice are initially comparably infected by Lm as measured by CFU assay at 1 hpi (Figure 3A). In P2+/+ PEMs the number of CFUs recovered at 6 hpi is 10-fold greater compared to the levels recovered at 1 hpi. Similar to previously published findings, in P2-/- PEMs the number of CFUs recovered at 6 hpi is 80-fold greater compared to the levels recovered at 1 hpi4. Enhanced Lm intracellular proliferation in P2-/- PEMs was accompanied with greater levels of viable Lm detected in the overlying media compared to levels detected in P2+/+ PEMs (Figure 3B). These data show that in an in vitro assay that the appearance of emergent extracellular Lm can also report on host factors that impact infection.
In vivo infection and analysis of the liver
In addition to the in vitro infection assays described above, the relative infectivity of the wild-type, attenuated, and hyper-virulent Lm strains can also be assessed by in vivo infection assays. Mice were intravenously co-infected with an equal mixture of the wild-type and ΔprfALm strains and 18 h later were humanely euthanized, livers excised, and single cell preparations were made. Isolated liver cells were lysed and the resulting lysates subjected to a CFU assay that could distinguish between the wild-type and ΔprfALm strains. Compared to the 1:1 ratio of these two strains in the original inoculum (the 'input'), the ΔprfA/wild type ratio at 18 hpi (the 'output') was considerably less than unity (for 6 mice range was 0.001 to 0.330; mean 0.100) (Figure 4). In contrast, mice co-infected with an equal mixture of the wild-type and prfA*Lm strains the prfA*/wild type ratios ranged from 2.4 to 10.2 (mean 7.1). This experiment shows that the relative infectivity of these three Lm strains (wild type, ΔprfA, and prfA*) in an intact organism resembles that observed in in vitro infection assays.
The host can also be monitored to determine whether the varying level of infectivity of the wild-type, ΔprfA, and prfA* strains are associated with a corresponding gradation of the innate immune response. Previously it has been shown that resident macrophages (Küpffer cells) and infiltrating inflammatory monocytes and neutrophils play critical and interrelated roles in protecting the liver during Lm infection5,6,7,8. Cells prepared from uninfected mice and mice infected with the GFP-expressing wild-type Lm strain for 18 h were stained with cell type-specific markers and analyzed by flow cytometry. In terms of non-immune cells of the liver (e.g., hepatocytes and endothelial cells), there were no detectable differences between uninfected and infected mice. In contrast, there were notable infection-related changes in the liver in terms of the proportion of immune cells that positively stained for the leukocyte common antigen CD45 (Figure 5A). Specifically, mice infected with the wild-type and prfA*Lm strains had significantly higher levels of CD45+ cells in the liver compared to uninfected mice whereas the levels of CD45+ liver cells in mice infected with the attenuated ΔprfALm strain were not notably different than levels observed in uninfected mice. To further characterize both resident and infiltrating immune cell populations, CD45+ cells can be furthered subtyped. As observed by others9, a notable shift with infection is the almost complete disappearance of resident Küpffer Cells (CD11bmid F4/80+) in the liver and the concomitant appearance of a distinct population of CD11bhi cells that represent infiltrating neutrophils (Figure 5B; lower middle panel). Also appearing in the liver at 18 hpi, are CD11bmid F4/80– cells appear that, since they also are Ly6Chi, represent infiltrating inflammatory monocytes (Figure 5B; lower middle and right panels). In infected mice these latter cells are the only cells in which appreciable GFP signal was detected following 18 hs of infection (Figure 5C). This latter finding is similar to that recently reported by Jones and D'Orazio who showed that in mice, Lm primarily is associated with Ly6Chi monocytes that infiltrate the gut following food-borne infection10. The profile of liver CD45hi cells of mice infected with the Lm ΔprfA strain resembled that of uninfected mice whereas mice infected with the hyper-virulent LmprfA* strain had modestly enhanced levels of infiltrating neutrophils and inflammatory monocytes compared to mice infected with the wild-type Lm strain (data not shown).
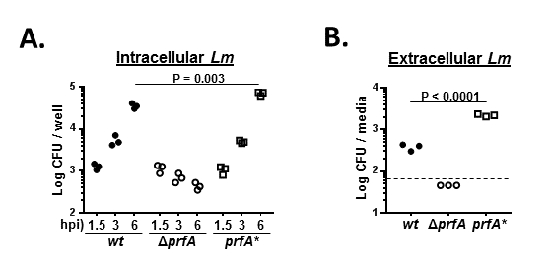
Figure 1: Infection of cultured macrophages by Listeria monocytogenes (Lm). The mouse macrophage cell line RAW 264.7 were infected in vitro with either wild-type Lm (wt; filled circles), attenuated Lm (ΔprfA; open circles), or hyper-virulent (prfA*; open squares) Lm strains (see text for details). (A) At 1.5, 3, and 6 hours post infection (hpi), macrophages were lysed and the cellular contents were analyzed by colony-forming unit (CFU) assay. (B) Infected macrophages were briefly treated with the antibiotic gentamicin to eliminate non-internalized Lm and at 6 hpi the overlaying media was collected and analyzed by CFU assay. Dotted line represents the detection limit of the assay. Each data point represents an independent well/infection and P values were determined by Student's t test. Shown are the results of a single representative experiment performed three times with similar outcomes. Please click here to view a larger version of this figure.
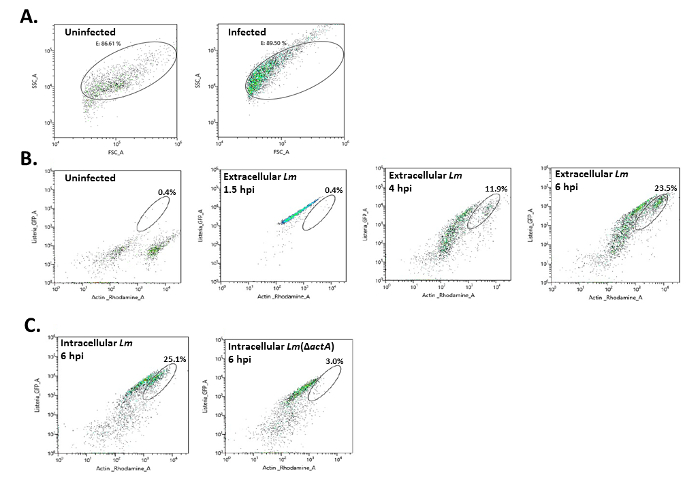
Figure 2: Actin-bound Lm analyzed by flow cytometry. The mouse macrophage cell line RAW 264.7 were either left uninfected or infected in vitro with GFP-expressing Lm. The overlying media was removed, centrifuged, and the collected material stained with actin-binding phalloidin. The stained material was analyzed by flow cytometry and shown is the light-scattering plots (A) indicating the gating used for the GFP- and phalloidin-associated levels of extracellular (i.e., media-derived) Lm shown in (B). (B) The indicated area represents the GFP/actin double positive Lm and their abundance as a percentage of the total events within the light-scattering gates. (C) Mouse macrophages were infected with either GFP-expressing Lm or a GFP-expressing Lm strain lacking the gene encoding ActA (ΔactA) that recruits the actin-polymerization machinery of the host cell to the Lm cell surface. At 6 hpi, infected cells were lysed and the released cellular contents, including the intracellular Lm, were stained for actin and analyzed by flow cytometry. Shown are the results of a single representative experiment performed three times with similar outcomes. Please click here to view a larger version of this figure.
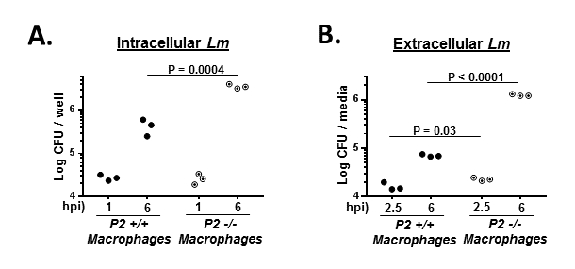
Figure 3: Infection of wild-type and Perforin-2 knockout macrophages by Lm. Peritoneal macrophages isolated from either a wild-type (P2+/+; filled circles) or knockout (P2-/-; dotted circles) were infected in vitro with Lm. (A) At 1 and 6 hpi, macrophages were lysed and the cellular contents analyzed by CFU assay. (B) Infected peritoneal macrophages were briefly treated with the antibiotic gentamicin and at 2.5 and 6 hpi the overlaying media was collected and analyzed by CFU assay. Each data point represents an independent well/infection and P values were determined by Student's t test. Shown are the results of a single representative experiment performed three times with similar outcomes. Please click here to view a larger version of this figure.
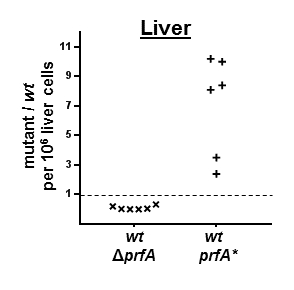
Figure 4: Lm infection of the mouse. Groups of 6 mice were infected with an equal mixture of either the wild-type Lm (wt) and attenuated Lm (ΔprfA) strains or an equal mixture of the wild-type Lm and hyper-virulent (prfA*) strains. At 18 hpi mice the Lm burden in the liver was determined by CFU assay. In this experiment the wild-type Lm strain possesses an antibiotic-resistance gene that allows it to be distinguished from the antibiotic-sensitive Lm mutant strains. Liver homogenates were plated on both antibiotic-containing and antibiotic-free media and the ratio of mutant and wild-type Lm were plotted. The dotted line represents the mutant/wild-type ratio of the infecting dose. The absolute levels of the wild-type Lm in the two cohorts of mice were 12 and 15 CFU/106 liver cells, respectively. Shown are the results of a single representative experiment performed two times with similar outcomes. Please click here to view a larger version of this figure.
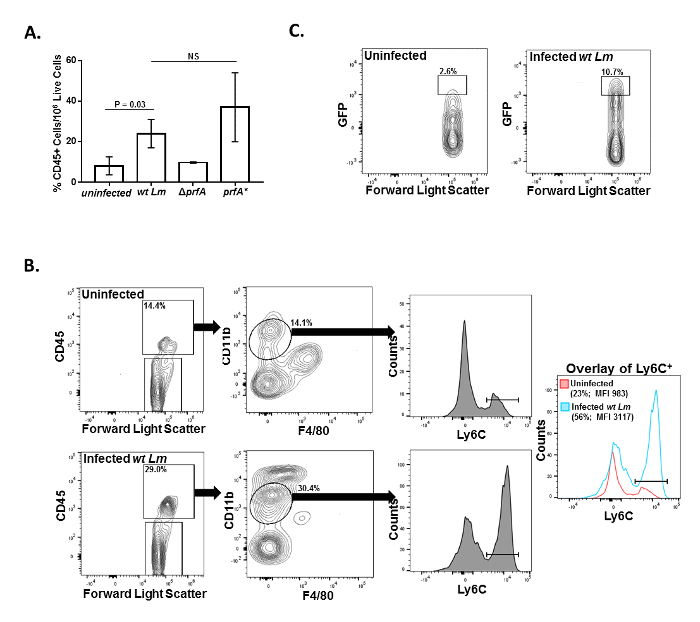
Figure 5: Immunological response to Lm infection in the mouse liver. Mice were either uninfected or systemically infected with 2 x 105 CFU of GFP-expressing Lm strains for 18 h. Single-cell preparations of livers were analyzed by flow cytometry for expression of immune- and myeloid-specific markers. (A) The percentage of cells staining positive for the immune-specific cell surface marker CD45+ per 106 isolated total live liver cells shown for uninfected mice and mice infected with either the wild-type (wt Lm), attenuated (ΔprfA), or hyper-virulent (prfA*) Lm strains. Plotted is the mean of three mice per group and P values were determined by Student's t test. (B) Shown in the left panels are the staining profile of CD45-staining liver cells from an individual uninfected mouse (top) and an individual wt Lm– infected (bottom); in the middle panels is shown the expression levels of myeloid-specific markers CD11b and F4/80 of the CD45+ cells; and on the right panels is shown the Ly6C expression levels of the indicated gated cells. In the far right panel is shown an overlay of the Ly6C+ cells from the uninfected and infected mice including the percentage and mean fluorescence intensities (MFI) of the CD11b+ F4/80– cells positive for the indicated Ly6C gate. Shown is a representative mouse from 3 mice per group. (C) Ly6C+ cells derived from livers of uninfected and infected mice (described (B); middle and right panels) were analyzed for GFP expression by flow cytometry. Please click here to view a larger version of this figure.
