The MXPressP workflow was used at the ESRF beamline MASSIF-1 to, fully automatically, mount, center in the X-ray beam, characterize, and collect full diffraction data sets from a series of crystals of human GCSH. The samples were mounted and the loop analyzed for an area to scan (Figure 1, left). After the diffraction analysis, four points were selected within the crystal for data collection (Figure 1, right). Subsequent processing by automated data analysis pipelines, including the MR pipeline, yielded high-quality datasets (Table 1) for which an MR solution was found. The latter allows users to rapidly evaluate whether the obtained dataset and the used search model are suitable for phasing by molecular replacement. In addition, the presence of ligands can be judged, thus permitting the user to focus only on the most promising datasets for further analysis. Manual structure determination by MR yielded a high-quality electron density map after a single automated refinement cycle (Figure 2a). For this dataset, the automated pipeline cut the data at a 1.32 Å resolution; however, users can still decide to cut the data at a lower resolution to arrive at different quality statistics (CC1/2, <I/σ(I)>, Rmeas) in the highest resolution shell. The crystal structure of human GCSH structure is similar to that of the bovine protein (3KlR)16.
Continuous electron density is visible for the entire amino acid chain, apart from the N-terminal histidine tag. Of the four substitutions that distinguish human and bovine GCSH, three are readily identifiable in the electron density (Ile/Val66, Asp/Glu98, and Leu/Phe149; Figure 2b-d). This is less clear for the Asp/Lys125 substitution for which the electron density of the side chain is only partially resolved due to flexibility (Figure 1e). The currently obtained model has Rwork and Rfree values of 20.4% and 23.8%, respectively, and can be further optimized by further cycles of automated and manual model building and refinement.
| GRENADES pipeline | XDS_APP pipeline | |
| Data collection and processing | ||
| X-ray source / Beam line | ESRF / MASSIF-1 | |
| Wavelength (Å) | 0.966 | |
| Resolution (Å) | 41.88 – 1.48 (1.53 – 1.48) | 41.86 – 1.32 (1.39 – 1.32) |
| Total/Unique reflections | 127670 / 28644 | 177332 / 40134 |
| (12178 / 2775) | (23772 / 5714) | |
| Space group for indexing, scaling and merging | C222 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 42.20, 83.75, 95.85 | 42.19, 83.72, 95,82 |
| Mosaicity | 0.05 | 0.05 |
| Rmeas (%) | 10.0 (110.7) | 11.1 (198.2) |
| <I/σ(I)> | 9.6 (1.3) | 7.6 (0.7) |
| CC1/2 (%) | 99.7 (53.9) | 99.7 (19.1) |
| Completeness (%) | 99.6 (99.6) | 99.5 (98.6) |
| Multiplicity | 4.5 (4.4) | 4.4 (4.2) |
| Molecular replacement and preliminary model refinement | ||
| Space group for phasing | C2 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 83.74, 42.18, 95.82 | 42.19, 83.72, 95,82 |
| α, β, γ (°) | 90, 90.03, 90 | 90, 90, 90 |
| Search model for MR (PDB) | 3KLR | 3KLR |
| Protein molecules / ASU | 2 | 1 |
| Protein residues | 250 | 125 |
| Rwork/Rfree (%) after 1st refinement | 24.3 / 26.5 | 20.4 / 23.8 |
| RMSD bond length (Å) after 1st refinement | 0.01 | 0.01 |
| RMSD bond angle (°) after 1st refinement | 1.2 | 1.83 |
| Rotamer outlier (%) after 1st refinement | 1.07 | 4.29 |
| Ramachandran favoured/allowed/disallowed (%) after 1st refinement | 95.93 / 4.07 / 0 | 95.12 / 4.88 / 0 |
Table 1: X-ray diffraction data collection, refinement, and validation statistics. Values for the highest resolution shell are given in brackets.
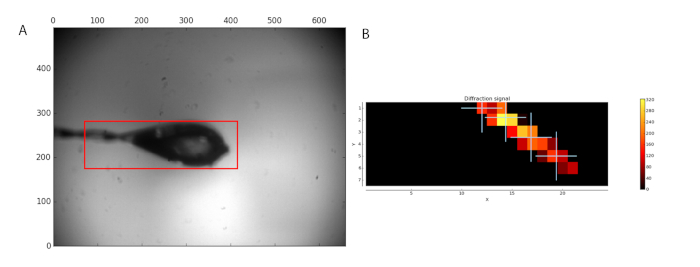
Figure 1: Sample analysis before data collection. (A) The region selected for scanning is shown by a red box. (B) The analysis of diffraction images is shown as a heat map. Four positions within the located crystal were selected for data collection. Please click here to view a larger version of this figure.
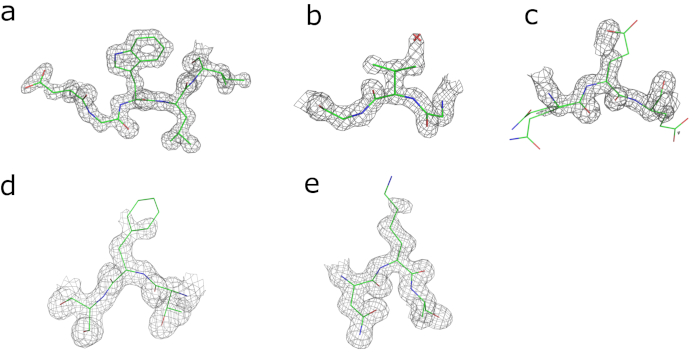
Figure 2: Visual validation of electron density maps obtained after refinement. Electron density maps contoured at 2x r.m.s. level around (a) Trp143, (b) Val66 (Ile in human GCSH), and (c) Glu98 (Asp in human GCSH) and maps contoured at 1x r.m.s level around (d) Phe149 (Leu in human GCSH) and (e) Lys125 (Asp in human GCSH). Please click here to view a larger version of this figure.
