The sc MHC class I technology used here allows precise control of cell surface expression of MHC class I with known, covalently linked peptides, to induce T cell activation. We have generated an engineered xenogeneic APC system (Figure 1A,B,C) that allows presentation of low levels of agonist pMHC in the presence or absence of co-agonist pMHC (Figure 1D,E). Critically, we have used a TCR-like antibody specific for HLA-A2 presenting E183 peptide to show that presentation of the agonist sc E183-HLA-A2 is not altered by co-expression of co-agonist sc GAG-HLA-A2 (Figure 1E). However, it must be noted that the E183-HLA-A2 specific TCR-like antibody shows some binding to HLA-A2 presenting GAG peptide (Figure 1E). Similar engineered antigen presenting cell systems can be constructed using different peptides or MHC molecules to investigate molecular requirements for TCR and/or coreceptor interactions in human T cells with different specificities.
We used these engineered APCs to stimulate an E183-HLA-A2-specific human CD8+ T cell clone. Three negative controls were used: T cells only, untransfected T-REx CHO cells, and T-REx CHO cells expressing high amounts of co-agonist sc GAG-HLA-A2 to test for potential T cell activation by any molecules expressed on T-REx CHO cells (untransfected T-REx CHO cells as compared to T cell only), and for T cell activation by sc-GAG-HLA-A2 (T-REx CHO cells expressing high levels of sc-GAG-HLA-A2 as compared to untransfected T-REx CHO cells). Untransfected T-REx CHO cells or T-REx CHO cells expressing sc-GAG-HLA-A2 did not induce activation of E183-HLA-A2-specific CD8+ CTL clone, as determined by quantifying IFN-γ production and expression of degranulation marker CD107a as a measure of T cell cytotoxicity (Figure 2A,B). Expression of high levels of agonist sc E183-HLA-A2, used as positive control, induced very efficient IFN-γ production and degranulation (Figure 2A,B), demonstrating that the sc HLA-A2 construct can be recognized by a specific TCR to activate T cell effector functions. Expression of low levels of sc E183-HLA-A2 induced lower levels of IFN-γ production and degranulation, and this was enhanced by presence of co-agonist sc GAG-HLA-A2 (Figure 2A,B). It must be noted that very high percentages of CD107a+ T cells were observed even in response to low levels of antigenic pMHC (Figure 2B), consistent with previous reports of relatively low requirements for the amount of agonist pMHC for induction of T cell cytotoxicity33. The CD107a MFI, indicating the amount of degranulation on a single cell basis, provides a better dynamic range (Figure 2B). The T cell activation assay used allows simultaneous quantification of two major effector functions: cytokine production and cytotoxicity, and provides very sensitive readout with good dynamic range.
We used the single chain technology to test whether TCR binding to co-agonist pMHC is required for co-agonist pMHC-dependent activation enhancement. We mutated valine at position 152 at the edge of the HLA-A2 peptide binding groove (Figure 3B) to glutamic acid to create V152E mutant, as this mutation has been reported to abolish TCR binding to HLA-A234. We then tested if the V152E mutation can abolish TCR binding for the E183-HLA-A2 CTL clone used, by introducing this mutation into agonist sc E183-HLA-A2 and expressing the mutant agonist sc construct in T-REx CHO cells. Wild type, but not V152E, sc E183-HLA-A2 induced activation of the E183-HLA-A2-specific CTL clone used (Figure 3A). It must be noted that any TCR-binding mutation on MHC class I needs to be separately tested for each individual TCR/T cell clone used, as the effect of mutation will depend on the precise molecular interactions between TCR and pMHC complex, and these will differ between different TCRs. For example, we tested eight mutations, either previously reported TCR-binding mutations or mutations predicted to disrupt TCR binding without abrogating peptide binding into HLA-A2. Of these, V152E was the only mutation that abolished activation of the E183-specific T cell clone used10. We then introduced the V152E mutation into the co-agonist sc GAG-HLA-A2, and co-expressed WT or V152E sc GAG-HLA-A2 with low levels of agonist sc E183-HLA-A2. Both WT and V152E sc GAG-HLA-A2 enhanced IFN-γ production and degranulation (Figure 3C,D), suggesting that TCR binding to co-agonist pMHC is not required for co-agonist pMHC-dependent CD8+ T cell activation enhancement. Single chain MHC class I technology, as presented here, can be used to modify TCR and/or coreceptor binding to MHC class I with known peptide to investigate the molecular requirements for T cell antigen recognition and activation.
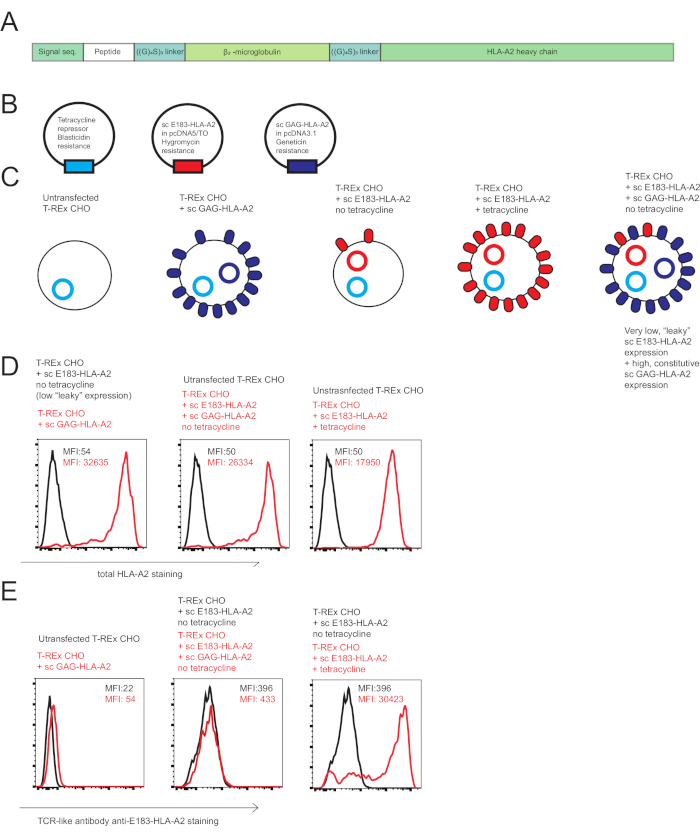
Figure 1: Single chain HLA-A2 constructs used to investigate co-agonist in human CD8+ T cell activation. (A) Schematic diagram of a scHLA-A2 construct, consisting of covalently signal sequence from fused human β2 microglobulin, peptide of choice, flexible glycine-serine linker (GGGGSGGGGS GGGGS), human β2 microglobulin, flexible glycine-serine linker and HLA-A2 heavy chain. (B) Schematic diagram of the plasmids used to generate engineered antigen presenting cells to investigate co-agonism in human CD8+ T cell activation: tetracycline repressor, agonist sc E183-HLA-A2 under control of tetracycline-inducible promoter, non-stimulatory co-agonist sc GAG-HLA-A2 under control of constitutively active promoter. (C) Schematic diagram of the engineered antigen presenting cells used to investigate co-agonism: T-REx CHO cells (no human MHC expression), T-REx CHO cells expressing constitutively high levels of sc GAG-HLA-A2 (non-stimulatory co-agonist pMHC), T-REx CHO cells expressing low “leaky” levels of sc E183-HLA-A2 in the absence of tetracycline (low level of agonist peptide), T-REx CHO cells expressing high levels of sc E183-HLA-A2 in the presence of tetracycline (high level of agonist peptide), T-REx CHO cells expressing low levels of sc E183-HLA-A2 and high levels of sc GAG-HLA-A2 (low level of agonist peptide and high level of co-agonist peptide). (D) Cell surface staining of the engineered antigen presenting cell lines using anti-HLA-A2 antibody. The numbers shown denote MFI values for MHC stain in the live cell gate. Data representative of 5 independent experiments (E) Cell surface staining of the engineered antigen presenting cell lines using TCR-like antibody specific against HLA-A2 presenting E183 peptide. The numbers shown denote MFI values for MHC stain in the live cell gate. Data representative of 5 independent experiments. Please click here to view a larger version of this figure.
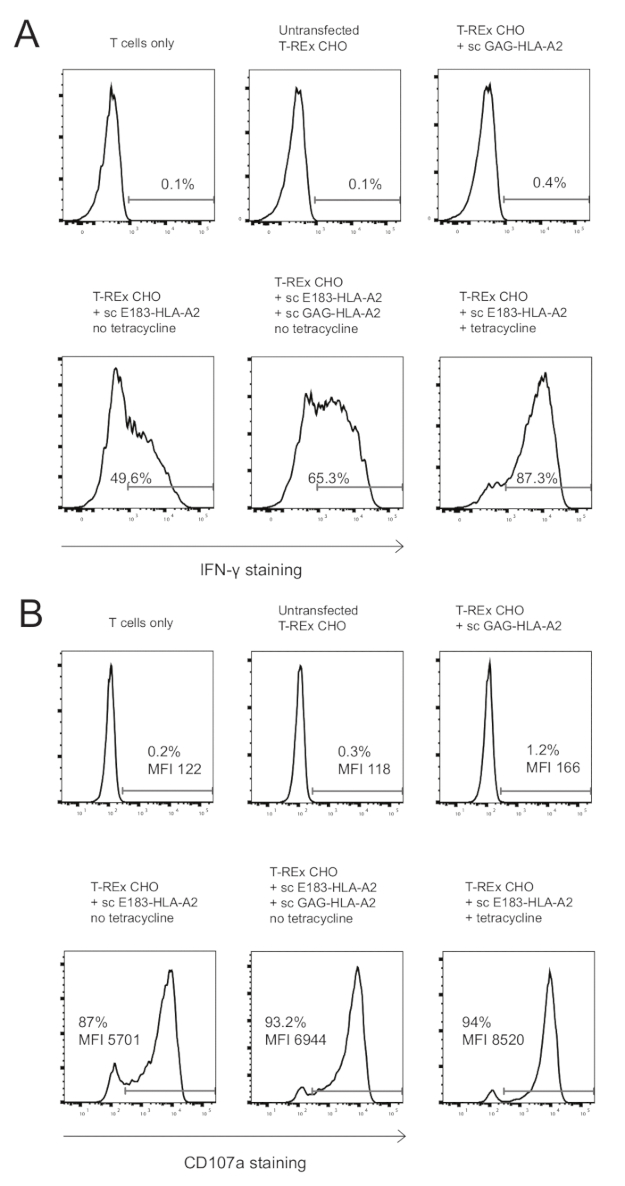
Figure 2: Presence of co-agonist pMHC enhanced cytokine production and degranulation of E183-HLA-A2-specific human CD8+ CTL clone. (A) Intracellular IFN-γ staining of HLA-A2-specific human CD8+ CTL clone after 3h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for IFN- γ staining. Data representative of 3 independent experiments, each performed with technical triplicates. (B) Cell surface CD107a staining of HLA-A2-specific human CD8+ CTL clone after 3h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for CD107a staining or the CD107a MFI for the CD8+ T cell population. Data representative of 3 independent experiments, each performed with technical triplicates. Please click here to view a larger version of this figure.
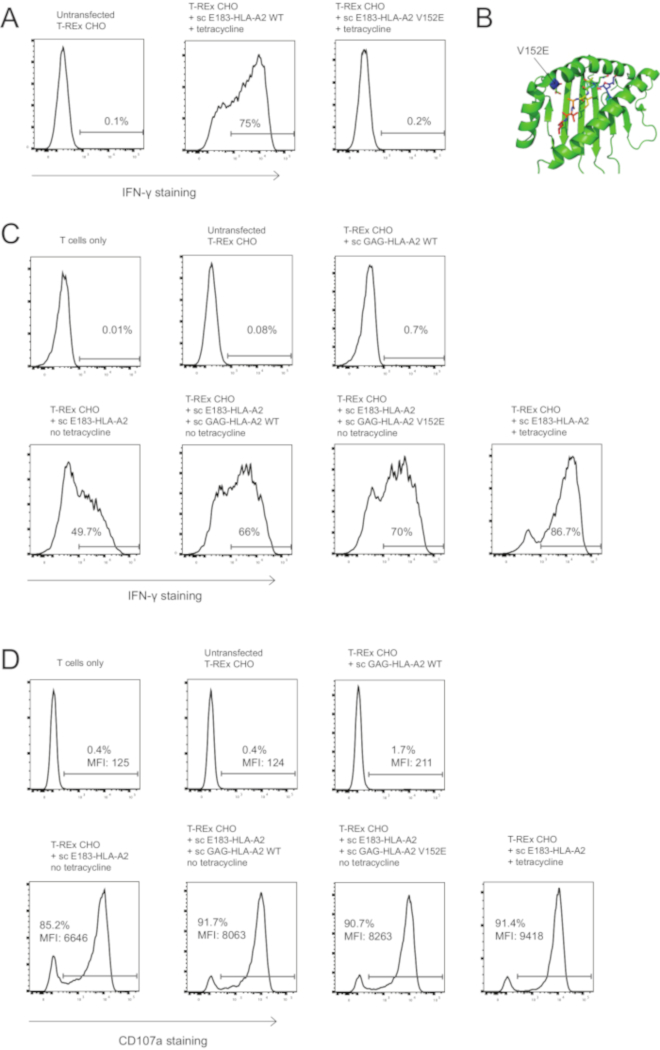
Figure 3: Co-agonist – mediated T cell activation enhancement does not require TCR binding to the co-agonist pMHC complex. (A) V152E mutation in sc E183-HLA-A2 abolishes activation of the E183-HLA-A2-specific human CD8+ CTL clone. T-REx CHO cells expressing high levels (tetracycline induction) of the WT or V152E mutant sc E183-HLA-A2 were used to stimulate E183-HLA-A2-specific human CD8+ CTL clone for 3h. T cell activation was quantified using intracellular IFN-γ staining. The numbers shown denote the percentage positive for IFN- γ staining. Data representative of 3 independent experiments, each performed with technical triplicates. (B) Location of V152E mutation within the peptide binding groove of HLA-A2. (C) V152E mutation on sc GAG-HLA-A2 co-agonist pMHC complex does not abolish co-agonist-dependent activation enhancement. Intracellular IFN-γ staining of HLA-A2-specific human CD8+ CTL clone after 4h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for IFN- γ staining. Data representative of 3 independent experiments, each performed with technical triplicates. (D) V152E mutation on sc GAG-HLA-A2 co-agonist pMHC complex does not abolish co-agonist-dependent activation enhancement. Cell surface CD107a staining of HLA-A2-specific human CD8+ CTL clone after 3h stimulation with the indicated engineered antigen presenting cells. The numbers shown denote the percentage positive for CD107a staining or the CD107a MFI for the CD8+ T cell population. Data representative of 3 independent experiments, each performed with technical triplicates. Please click here to view a larger version of this figure.
