Yolk volume reduction is important for normal morphology
As recently described in Almuedo-Castillo et al.17, size reduction of embryos can be achieved without reducing yolk volume. To compare with and without yolk volume reduction, we performed both two-step chopping (both blastula and yolk) and blastula-only chopping (Figure 2 and Supplemental Movie 1). Two-step chopped embryos showed seemingly normal overall morphology compared to the control (dechorionation only) embryos, other than size difference, throughout the developmental stages (see top and middle panels in Figure 2). On the other hand, blastula-only chopped embryos showed a peculiar morphology, especially at earlier stages. During epiboly, the embryos had a constricted and indented appearance (See bottom panel for 70% epiboly in Figure 2). At the following somite stage, midline structures were found to be flattened (i.e. DV length is relatively shorter than ML length) at many axial levels (See bottom panels for 8 and 20 somite in Figure 2). At later stages, the body structures adjacent to the yolk, such as the mid- and hindbrain, and the first ~10 somites, still showed a relatively flattened shape, possibly due to increased tension from the relatively larger yolk.
Somite size reduction in size reduced embryos
Somites are segmental structures that appear transiently during embryogenesis and give rise to vertebrae and skeletal muscle. From presomitic mesoderm (PSM), somites are formed one by one from the anterior to posterior direction in a periodic manner (e.g. 25 min for zebrafish, 2 h for mice) (Figure 3A). We performed time lapse imaging of somite formation both for control and chopped embryos and measured the size of most newly formed somites (Figure 3B). In both control and chopped embryos, the sizes of somites that were formed at later stages were found to be smaller compared to the ones from earlier stages. Also, throughout the somite formation stages, chopped embryos had smaller somites than the ones in control embryos (Figure 3C).
Neural tube heights are reduced following size reduction
To see the effect of embryo size reduction on neural tube size, we performed our two-step chopping technique on mem-mCherry injected embryos and imaged their spinal cords at 20 hpf using our confocal imaging system (Figure 4A,B). In this dataset, neural tube heights were reduced following size reduction by 12.4% ± 3.2%, as measured manually using custom image analysis code (Figure 4C). Taken together, these data show that size reduction reduces neural tube height. This technique can be used to measure the effects of size reduction on neural patterning.
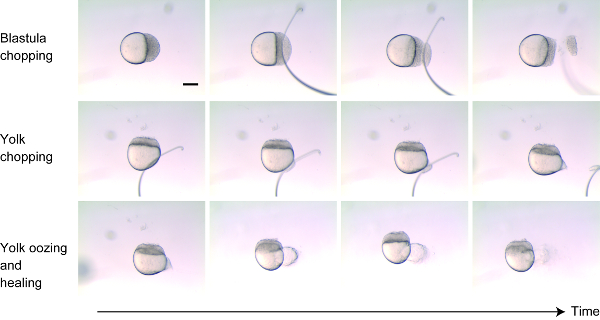
Figure 1: Size reduction technique. Approximately 30%-40% of the cells were cut from the animal pole (top panels). The membrane surrounding the yolk was carefully wounded so that the yolk oozed out (middle panels). For the following few minutes, yolk oozed out and then the wounds on both blastoderm and yolk healed up (bottom panels). Scale bar = 200 μm. Please click here to view a larger version of this figure.
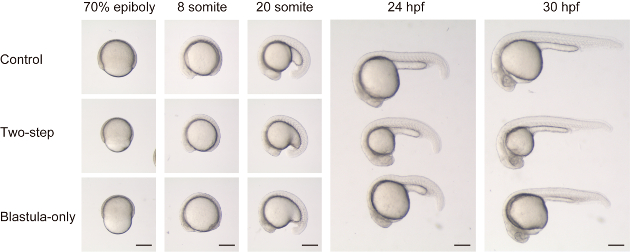
Figure 2: Comparison between two methods of size reduction. Control embryos (top panels, top embryos for 24 hpf and 30 hpf), size reduced embryos with two-step chopping (blastula and yolk, middle panels, middle embryos for 24 hpf and 30 hpf) and size reduced embryos with blastula-only chopping (bottom panels, bottom embryos for 24 hpf and 30 hpf) are compared along developmental stages. Note that in blastula-only chopped embryos, the blastoderm volume is much smaller compared to the yolk (at 70% epiboly). As a result, the embryo has a disproportionately flattened shape at somite stages (i.e., DV axis is relatively shorter compared to AP axis in blastula-only chopped embryos, compared to the control or a two-step chopped one). Scale ba = 200 μm. Please click here to view a larger version of this figure.
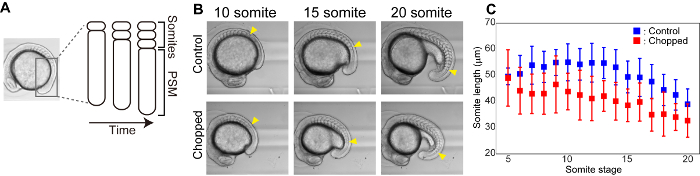
Figure 3: Size reduction reduces the length of somites. (A) Schematic illustration of somite formation. (B) Bright field images of control and chopped embryos over time. Yellow arrowheads indicate the most newly formed somite at each somite stage. (C) Somite length (in anterior-posterior axis) measurements over time for both control and chopped embryos. Error bars represent standard deviation. Please click here to view a larger version of this figure.
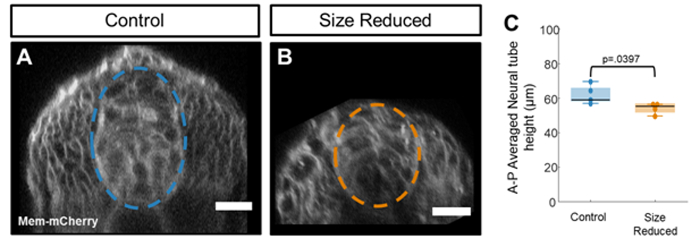
Figure 4: Size reduction reduces the height of the neural tube. (A-B) Example images of normal sized (A) and size reduced (B) tg(ptch2:kaede) embryos which were injected at the single cell stage with mem-mCherry mRNA. Scale bar = 20 μm. (C) Neural tube heights extracted from manual segmentation of the neural tube in each z-stack. Statistically significant differences are observed in average neural height when values are compared using an unpaired t-test (p = 0.0397). Please click here to view a larger version of this figure.

Supplemental Movie 1: Comparison between two-step chopping versus blastula-only chopping. Top row = control embryos, middle row = size reduced embryos with two step chopping, bottom row = size reduced embryos with blastula only chopping. Movies were taken every 3 min for 12 h. Scale bar = 1 mm. Please click here to view this video. (Right-click to download.)
