Radiosensitivity of Cancer Stem Cells in Lung Cancer Cell Lines
Summary
The presence of cancer stem cells have been associated with relapse or poor outcomes after radiotherapy. This manuscript describes the methods to study the radiosensitivity of cancer stem cells in lung cancer cell lines.
Abstract
The presence of cancer stem cells (CSCs) has been associated with relapse or poor outcomes after radiotherapy. Studying radioresistant CSCs may provide clues to overcoming radioresistance. Voltage-gated calcium channel α2δ1 subunit isoform 5 has been reported as a marker for radioresistant CSCs in non-small cell lung cancer (NSCLC) cell lines. Using calcium channel α2δ1 subunit as an example of a CSC marker, methods to study the radiosensitivity of CSCs in NSCLC cell lines are presented. CSCs are sorted with putative markers by flow cytometry, and the self-renewal capacity of sorted cells is evaluated by sphere formation assay. Colony formation assay, which determines how many cells lose the ability to generate descendants forming the colony after a certain dose of radiation, is then performed to assess the radiosensitivity of sorted cells. This manuscript provides initial steps for studying the radiosensitivity of CSCs, which establishes the basis for further understanding of the underlying mechanisms.
Introduction
Radiotherapy plays an important role in cancer treatment. However, existence of radioresistant cancer stem cells (CSCs) may lead to relapse or poor outcomes after radiotherapy1,2. CSCs are characterized by their self-renewal capacity and ability to generate heterogenous cancer cells3. Armored with a more efficient DNA damage repair capacity or higher levels of free-radical scavenging systems or other mechanisms, CSCs are relatively resistant to radiotherapy4,5,6,7,8. Identifying CSC markers and exploring their mechanisms will facilitate the development of drugs that will overcome radioresistance without increasing normal tissue damage.
Voltage-gated calcium channel α2δ1 subunit isoform 5 has been reported as a marker for radioresistant CSCs in NSCLC cell lines9. α2δ1 was originally identified as a CSC marker for hepatocellular carcinoma (HCC)10. Using subtractive immunization with a pair of HCC cell lines derived from the primary and recurrent tumors in the same patient, an antibody named 1B50-1 was identified to target recurrent HCC cells specifically. 1B50-1-positive cells showed high sphere formation efficiency in vitro and high tumorigenicity in vivo. Its antigen was identified by mass spectrometry, as calcium channel α2δ1 subunit isoform 5. α2δ1 specifically expresses in CSCs and is undetectable in most normal tissues, making it a potential candidate for targeting CSCs10. α2δ1 can also serve as a CSC marker for NSCLC cell lines, and it has been shown to impart radioresistance to NSCLC cells partially by enhancing the efficiency of DNA damage repair in response to radiation9.
Studying radioresistant CSCs may provide clues to overcoming radioresistance. Using α2δ1 in NSCLC as an example, major methods to study the radiosensitivity of CSCs are presented. Usually, CSCs are isolated with a putative surface marker, and the stem cell characteristics and radiosensitivity of the positive and negative cell populations are compared. Sphere formation in a serum-free medium supplemented with growth factors that support self-renewal is a useful assay to evaluate the stemness of cells in vitro. Cells with high sphere formation capacity are likely to show high tumorigenicity when injected into immunodeficient mice10,11,12. Colony formation assay is then used to assess the radiosensitivity of cells, which determines how many have lost the ability to generate descendants forming the colony after a dose of radiation13.
Protocol
NOTE: Steps are performed under the indicated temperature. For steps in which the temperature is not mentioned, perform under room temperature (18–25 °C). Cell culture medium should be stored at 4 °C, and other reagents should be stored according to the manufacturer’s guides. Medium should be pre-warmed to 37 °C before being added to cells.
1. Cell sorting
- Antibody conjugation
NOTE: Considering the shorter incubation time, direct-labeled antibody is preferred for cell sorting. If no commercial direct-labeled antibody is available, use fluorescein-conjugating reagents to conjugate the antibody to a fluorescent dye according to the manufacturer’s guide (see Table of Materials). As cells will be cultured after sorting, all steps should be performed in a biological safety cabinet or laminar clean bench. To avoid contamination, antibiotics (penicillin and streptomycin) are recommended to be added to the culture medium after sorting.- Add 1 µL of modifier reagent for each 10 µL of antibody to be labeled. Mix gently.
- Add the mixture of antibody and modifier directly onto the lyophilized fluorescein. Pipet gently. Leave the vial standing for 3 h or overnight at room temperature (RT) in the dark.
- Add 1 µL of quencher reagent for every 10 µL of antibody. The conjugate can be used after 30 min. The fluorescein conjugate can be stored at 4 °C for up to 18 months.
- Before sorting, titration of conjugated antibody is recommended. Prepare cells that express (e.g., H1299 or A549 for α2δ1) and do not express (e.g., H1975) the marker. Aliquot the cells and incubate them with antibody at different concentrations (e.g., 15 μg/mL, 7.5 μg/mL, 1.5 μg/mL, and 0.75 μg/mL, respectively).
- Perform flow cytometric analysis (with histogram or dot plot) and choose the concentration in which positive cells of H1299 or A549 can be separated from the isotype control and H1975 cells have little unspecific signals.
- Cell labeling
- Culture A549 cells in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) in 10 cm dishes at 37 °C and 5% CO2 in a cell culture incubator. Digest cells as follows for sorting when they reach 80%–90% confluence (about 5–10 x 106 cells per dish).
- Remove the culture medium. Wash cells with 3 mL of phosphate buffered saline (PBS) briefly. Add 3 mL of 0.05% trypsin to each dish. Remove the trypsin and leave the residuary trypsin to digest cells at 37 °C for 1–3 min. Check the detachment of the cells frequently to avoid overdigestion.
NOTE: Some cell lines may be relatively difficult to digest, such as NSCLC cell lines H520 and PC9. In such situation, 3 mL of trypsin can be left in the dish, rather than digestion with residuary trypsin. Moreover, the digestion time can be extended. - When cells become loose and begin to detach from the dishes, add 3 mL of RPMI 1640 medium supplemented with 10% FBS and pipette the cell suspension into a 50 mL tube.
- Centrifuge at 300 x g at 4 °C for 5 min. Discard the supernatant. Add 10 mL of PBS to resuspend the cells. Transfer 0.5 mL (or at least 5 x 105 cells) of cell suspension into a new tube as an isotype control. The remaining cells are for sorting.
- Centrifuge both the control and experimental tubes at 300 x g at 4 °C for 5 min. Discard the supernatant.
- Prepare the working solution for staining by diluting the fluorescent dye-conjugated isotype control or antibody in PBS to the optimal concentration titrated as mentioned above.
NOTE: In this experiment, FITC-conjugated 1B50-1 (the antibody of α2δ1) was diluted into 7.5 μg/mL. The volume of working solution is dependent on the cell amount. - Resuspend the control and experimental cells with each working solution at about 2–5 x 107 cells/mL and mix gently. Incubate the samples at RT for 30–40 min in the dark.
- Wash the cells by adding 10 mL of PBS to samples, mix gently and centrifuge at 300 x g at 4 °C for 5 min. Discard the supernatant. Resuspend cells with 10 mL of PBS.
- Put 40 μm cell strainers on new 50 mL tubes for control and experimental cells respectively. Apply cell suspension onto the strainer and collect the flow-through.
NOTE: This step removes the cell clumps that may block the capillary of the cell sorter. - Centrifuge the flow-through at 300 x g at 4 °C for 5 min. Discard the supernatant. Resuspend the cells with 0.2–1.0 mL PBS then transfer into a 5 mL tube for flow cytometry.
- Cell sorting by flow cytometry
- Prepare two 5 mL tubes to collect the positive and negative cell populations. Add 1 mL of RPMI 1640 medium in each tube. Place the tubes at the collection platform of the cell sorter.
- Analyze the control cells and experimental cells respectively with the dot plot. Gate the live cells with the parameters forward scatter (FSC) and side scatter (SSC). Gate the positive and negative population of live cells according to the FL-1 intensity.
- Collect the α2δ1-positive cells and α2δ1-negative cells. Record the number of cells obtained.
- To confirm that the harvested cell populations have different level of α2δ1 expression, a quantitative polymerase chain reaction (QPCR) can be performed.
- Extract RNA of positive and negative cells with guanidinium thiocyanate reagent and reverse transcript the RNA into cDNA according to the manufacturer’s guide. Perform QPCR with SYBR green reagent. The sequences of primers are as follows: α2δ1-F, CAGTTGAGATGGAGGATGATG; α2δ1-R, TTGTATGAGCAGTCGTGTGTC; GAPDH-F, GTCGGAGTCAACGGATTTGG; and GAPDH-R, AAAAGCAGCCCTGGTGACC.
2. Sphere formation assay
NOTE: Sphere formation assay is applied to determine the self-renewal capacity of cells. Cells with self-renewal capacity could form spheres in this serum-free semisolid medium supplemented with growth factors. All steps should be performed in a biological safety cabinet or laminar clean bench. To avoid contamination, antibiotics (penicillin and streptomycin) are recommended to be added to the culture medium after sorting.
- Medium preparation
- Prepare 2x DMEM/F-12 medium by reconstituting a 1 L package of DMEM/F-12 powder into 475 mL of distilled water. Add 2.438 g of NaHCO3. Adjust the pH to 7.0–7.2 by slowly adding 1 N NaOH or 1 N HCl with stirring. Add distilled water to a final volume of 500 mL. Sterilize the medium by filtering through a 0.2 μm filter.
- Prepare a 2% methylcellulose solution by adding 2 g of methylcellulose into 100 mL of distilled water. Autoclave the solution. The methylcellulose may show a cotton-like state after autoclaving. Mix it by reversing the bottle up and down gently for few times and leave it for natural cooling. It will become a transparent semisolid overnight.
- To obtain 20 mL of self-renewal medium, add 10 mL of 2x DMEM/F-12 medium to a 50 mL tube. Add 400 μL of B27 to the medium. Add recombinant human epidermal growth factor (EGF) to a final concentration of 25 ng/mL. Add recombinant human basic fibroblast growth factor (bFGF) to a final concentration of 25 ng/mL. Add 2% methylcellulose to reach a final volume of 20 mL.
- Mix the medium by vortex so that the self-renewal medium is obtained.
- Cell seeding
- Transfer the harvested cells into the self-renewal medium to reach 2000 cells/mL and mix them with a brief vortex. Add 100 μL of cell suspension to each well of an ultra-low attachment 96 well plate. Do not use the wells at the rim of the plate, as the medium is more likely to evaporate in these wells. Add sterilized water to these wells.
- Culture at 37 °C with 5% CO2 in cell culture incubator for 10–14 days. Add 50 μL of self-renewal medium at day 5–7.
- Sphere counting and calculation
- 10–14 days after seeding, count the number of spheres with more than 50 cells (approximately) or with a diameter more than 100 μm under a microscope (4x objective lens, 10x eyepiece).
NOTE: Starting from the upper left of the well, count the spheres while moving the objective table horizontally towards the right with the joystick of the microscope. Then, move the objective table to the middle row of the visual field and count the spheres from right to left. Then, move to the lower row and count from left to right. A well of 96 well plate can be counted within three rows. - Calculate the sphere formation efficiency as the number of spheres divided by the number of cells seeded. Compare the sphere formation efficiency between the positive and negative cell populations.
- 10–14 days after seeding, count the number of spheres with more than 50 cells (approximately) or with a diameter more than 100 μm under a microscope (4x objective lens, 10x eyepiece).
3. Colony formation assay
NOTE: Colony formation assay is applied to determine the radiosensitivity of cells. Radiation can be delivered by a linear accelerator used for radiotherapy. Cell irradiation system for laboratory use can also be used if the equipment is available. Steps 3.1, 3.2.3, and 3.2.6 should be performed in a biological safety cabinet or laminar clean bench. To avoid contamination, antibiotics (penicillin and streptomycin) are recommended to be added to the culture medium after sorting.
- Cell seeding
- Prepare one 6 well plate for each radiation dose including the 0 Gy group. Add 1.5 mL of medium to each well of 6 well plate. Culture medium for colony formation assay is the conventional RPMI 1640 medium supplemented with 10% FBS (named as “completed 1640”).
- Dilute harvested cells with completed 1640 medium to 1000 cells/mL.
- Add cell suspension to the wells. Shake the plates horizontally to make cells distribute evenly in the wells. Record the volume added in each dose group.
NOTE: Generally, seed 200 cells per well for 0 Gy, 200 cells for 2 Gy, 400 cells for 4 Gy, 800 cells for 6 Gy, and 1600 cells for 8 Gy, respectively. It is better to adjust the cell numbers in unsorted cells in pre-experiments. - Culture at 37 °C with 5% CO2 in cell culture incubator.
- Radiation
- After cells have attached to the bottom of the wells (usually one day after seeding, and longer time is not recommended considering cell proliferation), proceed to perform cell radiation.
- Calculate the number of monitor units (MU) for each dose at a depth of 1 cm in a 20 cm x 20 cm radiation field.
NOTE: Number of MU = dose (cGy)/output factor for the radiation field/percentage depth dose (PDD) for the depth. For example, the output factor for a 20 cm x 20 cm field is 1.066, PDD for 1.0 cm is 0.987, and number of MU for 8 Gy (800 cGy) is 760 (800/1.066/0.987 = 760.35). If multiple plates need to be radiated for each dose, a larger field can also be used, and the numbers of MU need to be recalculated according to the size of the field. Output factor and PDD differ in different accelerators. Refer to radiation physicists for these parameters. - Add completed 1640 to each well to reach 1 cm for the height of the medium.
NOTE: Cell growth area of 6 well plate can be found on the manufacturer’s guide. For example, if the growth area is 9.5 cm2 for each well of 6 well plate, and 1.5 mL of medium and 0.4 mL of cell suspension has been added on the day before, then add 7.6 mL medium to the well. The height is needed for dose build-up. A medium height of less than 0.8 cm is not recommended.
NOTE: Keep the plates covered during radiation. When the plates are transferred between cell culture room and radiation therapy room, wrap the plates with aluminum foil or put them in a clean box to avoid contamination. Hold the plates flat to avoid medium spilling out. - Set a 20 cm x 20 cm radiation field. Place a tissue-equivalent bolus of 1.0 cm thickness on the treatment couch, and place the 6 well plate on the bolus to keep them in contact. Make sure the whole plate is within the radiation field. Set the source skin distance as 100 cm. Align the medium surface level to the laser level by adjusting the height of the treatment couch.
NOTE: Plates with the same radiation dose can be radiated at the same time. For this case, a larger radiation field is needed, and the number of MU should be recalculated according to the corresponding output factor. Make sure all the plates are within the radiation field. - Deliver the each assigned dose to each plate in sequence.
CAUTION: The linear accelerator can only be operated by qualified technicians. Keep away from radiation. - Take the plates back to the cell culture room. Change the medium with 2 mL of completed medium for each well. Culture at 37 °C with 5% CO2 in cell culture incubator. Change the medium every 3–5 days.
- Staining
- 7–10 days after radiation, when many colonies form and before they grow too large and merge together, then perform staining and counting as follows. Remove the medium and wash briefly with PBS. Add 1 mL of 4% formaldehyde to each well to fix the cells.
CAUTION: Formaldehyde is volatile. Use formaldehyde in a fume hood. - After 10 min of fixation, remove the formaldehyde and wash with 2 mL of distilled water each well twice.
- Add 1 mL of 1% crystal violet stain solution to each well. Stain for 10 min. Remove the crystal violet stain solution, and wash with distilled water 3 times. Remove the water and dry the plates.
- 7–10 days after radiation, when many colonies form and before they grow too large and merge together, then perform staining and counting as follows. Remove the medium and wash briefly with PBS. Add 1 mL of 4% formaldehyde to each well to fix the cells.
- Colony counting and calculation
- Count the number of colonies with more than 50 cells. Do not include small colonies with less than 50 cells. Check the colonies under a microscope to get an impression of a colony constituted by 50 cells and mark it on the bottom of the plate with a marker pen.
NOTE: Photographing the wells and using the software ImageJ will facilitate the counting process. - Calculate the colony formation efficiency as the number of colonies divided by the number of cells seeded. Calculate the survival fraction as the colony formation efficiency at a certain irradiation dose divided by the colony formation efficiency at 0 Gy.
- Using the dose as the x-axis and survival fraction as the y-axis, a survival curve can be obtained. Compare the survival curves between the positive and negative cell populations.
- Count the number of colonies with more than 50 cells. Do not include small colonies with less than 50 cells. Check the colonies under a microscope to get an impression of a colony constituted by 50 cells and mark it on the bottom of the plate with a marker pen.
Representative Results
α2δ1-high and α2δ1-low A549 cells were sorted (Figure 1A). Some markers may show distinct populations and are easy to gate. However, some markers just show high and low expression patterns, rather than distinct positive and negative populations. In this situation, an isotype control is very important for gating. The expression of α2δ1 in sorted cells is validated by qPCR. The expression of CACNA2D1, the gene that encode α2δ1, is higher in sorted α2δ1-high cells compared with α2δ1-low cells (Figure 1B).
Typical morphology of spheres is shown in Figure 2A. The sphere formation efficiency is calculated in α2δ1-high and α2δ1-low cells (Figure 2B). α2δ1-high cells showed higher sphere formation efficiency, suggesting a higher self-renewal capacity. Typical images of cell colonies are shown in Figure 3A. A colony with about 50 cells can be examined under a microscope and marked as a reference. A survival fraction at each dose can be calculated, and the survival curves are presented in Figure 3B. α2δ1-high cells are relatively resistant to radiation compared to α2δ1-low cells.
Unpaired two-sided Student’s t-test was performed to evaluate the significance between groups. A value of p < 0.05 was considered to be statistically significant. Data are represented with the mean and standard deviation (SD). Representative data from at least three biologically independent experiments with similar results are presented.
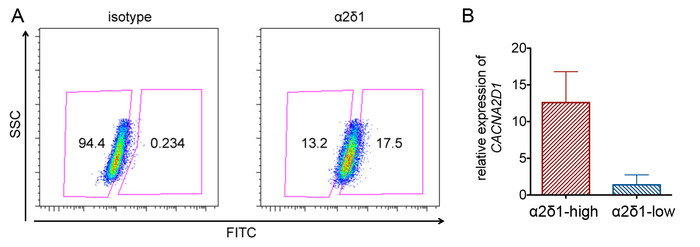
Figure 1: Sorting α2δ1-high and α2δ1-low cells in A549. (A) Representative flow cytometry analysis of α2δ1 expression. Cells are gated based on the isotype control. (B) Confirmation of α2δ1 expression in high and low populations by QPCR. The error bars indicate SD. Please click here to view a larger version of this figure.
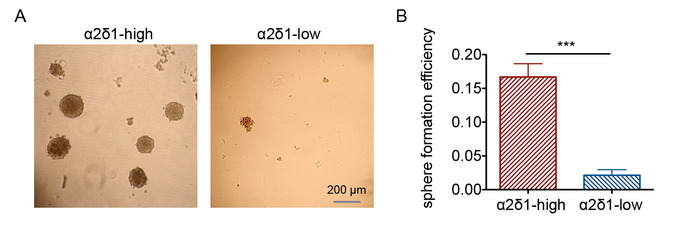
Figure 2: Sphere formation assay of α2δ1-high and α2δ1-low A549 cells. (A) Representative morphology of the spheres formed by the sorted α2δ1-high and α2δ1-low cells (bar = 200 μm). (B) Sphere formation efficiency of α2δ1-high and α2δ1-low cells. The error bars indicate SD (***p < 0.001). Please click here to view a larger version of this figure.
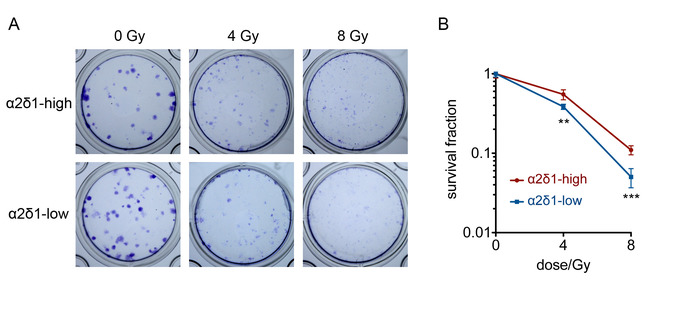
Figure 3: Colony formation assay of α2δ1-high and α2δ1-low A549 cells. (A) Representative images of the colonies formed by the sorted α2δ1-high and α2δ1-low cells. (B) Survival curves of α2δ1-high and α2δ1-low cells. The numbers of seeded cells were 200 cells per well for 0 Gy, 400 cells per well for 4 Gy, and 800 cells for 8 Gy. The error bars indicate SD (**p < 0.05, ***p < 0.001). Please click here to view a larger version of this figure.
Discussion
This protocol describes methods to study the radiosensitivity of CSCs in cancer cell lines in vitro. In this study, the expression of α2δ1 is continuous in NSCLC cell lines. Therefore, gating is based on an isotype control. Before sorting, α2δ1 expression should be examined in multiple cell lines by flow cytometry and validated by QPCR or western blot. It is recommended to re-analyze α2δ1 expression of the sorted α2δ1-high and α2δ1-low cells by flow cytometry, by observing fluorescence under a fluorescent microscope, or by QPCR (after sorting).
Sphere formation assay is a simple way to evaluate self-renewal capacity, which can be used to preliminarily characterize putative CSCs. This method has been used to culture neurospheres or mammospheres to characterize stem cells in glioma or breast cancer cells, and the formula is modified to culture other types of cancer4,6,10. EGF and bFGF can support the self-renewal of stem cells in serum-free medium; however, basic medium and growth factors may differ for culturing different cell types. The semisolid medium is used to immobilize cells so that spheres are formed by cell proliferation rather than clustering together. Ultralow attachment plate is used in the experiment to maintain sphere morphology. Moreover, as CSCs are enriched after sphere culture, when the spheres are collected, digested, and seeded for secondary sphere formation, sphere formation efficiency is likely to increase in subsequent serial propagation9,10. A more rigorous criteria for characterizing CSCs is in vivo limiting dilution assay, in which a series of numbers of sorted positive and negative cells are injected subcutaneously into immunodeficient mice, then the frequencies of tumorigenic cells in positive and negative cell populations are calculated10,14. If a putative cancer stem cell marker is identified by sphere formation assay, further characterization by in vivo limiting dilution assay is recommended.
In this study the radiosensitivity of CSCs is evaluated. Colony formation assay is a classical way to assess the radiosensitivity. Seeding same number of cells in parallel wells in important. Adjust the cell number per mL to a suitable range to ensure the volume of cell suspension added to each well is more than 100 μL. If the volume is too small, the error is likely to increased. For cell radiation, medium with a 1 cm height is added for dose build-up, and a tissue-equivalent bolus is placed under the plate for dose backscatter. Researchers are recommended to refer to radiation physicists and technicians to set up the cell radiation model and calculate the dose.
This manuscript provides the initial few steps for the radiosensitivity study of CSCs. Further mechanism studies may involve proteins related to DNA damage repair, clearance of reactive oxygen species, cell cycle arrest, etc.15. These experiments need a large amount of cells; therefore, many dishes of cells are needed to be prepared. Overexpression or knockout/knockdown of CSC genes also provide important insights into how CSCs gain radioresistance.
These methods can potentially be applied to identify CSCs in cancer tissues. Zhang et al. described isolating CD166-positive Lin-negative cells from NSCLC surgical samples12. Tissues were chopped with a sterile blade and digested with an enzyme cocktail. Cells collected by passing through cell strainers were labeled for sorting, and then characterized by sphere formation assay and in vivo limiting dilution assay. For α2δ1 as a CSC marker, it has been reported to isolate CSC in hepatocellular carcinoma surgical tissues and small cell lung cancer patient-derived xenograft tissues10,16. Considering a high number of cells are required for analysis, it is relatively difficult to isolate CSCs in biopsies or circulating tumor cells. Alternatively, staining the sections of biopsies may potentially provide clues for the existence of CSC in the tumors. However, this presumptive application needs to be evaluated in patient tissues and clinical data.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81402535 and 81672969) and National Key Research and Development Project (2016YFC0904703).
Materials
| 0.5% Trypsin-EDTA (10X), no phenol red | Thermo Fisher | 15400054 | Dilute in to 0.05% (1X) with autoclaved distilled water |
| 1B50-1 | This antibody is produced and friendly supplied by Laboratory of Carcinogenesis and Translational Reseach (Ministry of Education/Beijing), Department of Cell Biology, Peking University Cancer Hospital and Institute. See reference 10. Alternatively, commercial antibody of calcium channel α2δ1 subunit can be used (ABCAM, ab2864) (Yu, et al., Am J Cancer Res, 2016; 6(9): 2088-2097) | ||
| 4% formaldehyde solution | Solarbio | G2160 | |
| A549 | ATCC | RRID: CVCL_0023 | |
| B27 | Thermo Fisher | 17504044 | |
| Biological Safety Cabinet | Thermo Fisher | 1336 | |
| Centrifuge | Eppendorf | 5910R | |
| DMEM/F-12 | Thermo Fisher | 12500062 | |
| EGF Recombinant Human Protein | Thermo Fisher | PHG0311 | |
| Fetal bovine serum | Thermo Fisher | 16140071 | |
| FGF-Basic (AA 1-155) Recombinant Human Protein | Thermo Fisher | PHG0261 | |
| Flow cytometer/cell sorter | BD | FACSARIA III | |
| H1299 | ATCC | RRID: CVCL_0060 | |
| H1975 | ATCC | RRID: CVCL_1511 | |
| Lightning-Link Fluorescein Kit | Innova Biosciences | 310-0010 | |
| linear accelerator | VARIAN | CLINAC 600C/D | |
| Methyl cellulose | Sigma Aldrich | M7027 | |
| Penicillin-Streptomycin, Liquid | Thermo Fisher | 15140122 | |
| Phosphate buffered saline | Solarbio | P1020 | |
| RPMI-1640 | Thermo Fisher | 11875093 | |
| SYBRGREEN | TOYOBO | QPK-201 | |
| TRIzol | Thermo Fisher | 15596026 | |
| Violet crystal staining solution | Solarbio | G1062 |
Referencias
- Brunner, T. B., Kunz-Schughart, L. A., Grosse-Gehling, P., Baumann, M. Cancer stem cells as a predictive factor in radiotherapy. Seminars in Radiation Oncology. 22 (2), 151-174 (2012).
- Baumann, M., Krause, M., Hill, R. Exploring the role of cancer stem cells in radioresistance. Nature Reviews Cancers. 8 (7), 545-554 (2008).
- Clarke, M. F., et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Investigación sobre el cáncer. 66 (19), 9339-9344 (2006).
- Bao, S., et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444 (7120), 756-760 (2006).
- Wang, W. J., et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Investigación sobre el cáncer. 73 (3), 1219-1231 (2012).
- Diehn, M., et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 458 (7239), 780-783 (2009).
- Gomez-Casal, R., et al. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Molecular Cancer. 12 (1), 94 (2013).
- Mihatsch, J., et al. Selection of radioresistant tumor cells and presence of ALDH1 activity in vitro. Radiotherapy and Oncology. 99 (3), 300-306 (2011).
- Sui, X., Geng, J. H., Li, Y. H., Zhu, G. Y., Wang, W. H. Calcium channel α2δ1 subunit (CACNA2D1) enhances radioresistance in cancer stem-like cells in non-small cell lung cancer cell lines. Cancer Management and Research. 10, 5009-5018 (2018).
- Zhao, W., et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel α2δ1 subunit. Cancer Cell. 23 (4), 541-556 (2013).
- Moncharmont, C., et al. Targeting a cornerstone of radiation resistance: Cancer stem cell. Cancer Letters. 322 (2), 139-147 (2012).
- Zhang, W. C., et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 148 (1-2), 259-272 (2012).
- Franken, N. A., Rodermond, H. M., Stap, J., Haveman, J., van Bree, C. Clonogenic assay of cells in vitro. Nature Protocols. 1 (5), 2315-2319 (2006).
- O’Brien, C. A., Kreso, A., Jamieson, C. H. Cancer stem cells and self-renewal. Clinical Cancer Research. 16 (12), 3113-3120 (2010).
- Morgan, M. A., Lawrence, T. S. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clinical Cancer Research. 21 (13), 2898-2904 (2015).
- Yu, J., et al. Mechanistic exploration of cancer stem cell marker voltage-dependent calcium channel α2δ1 subunit-mediated chemotherapy resistance in small-cell lung cancer. Clin Cancer Res. 24 (9), 2148-2158 (2018).
